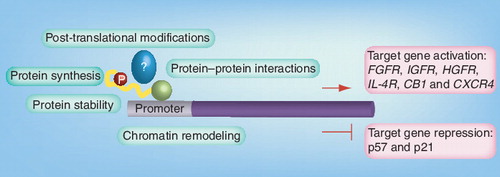
Only selected target genes are indicated. PAX–FKHR is shown in yellow/green, possible interaction partners are largely unknown (blue) and only phosphorylation (red) is indicated as an example for post-translational modification.
CB1: Cannabinoid receptor 1; CXCR4: CXC motive chemokine receptor; FGFR: FGF receptor; HGFR: HGF receptor; IGFR: IGF receptor; IL-4R: IL-4 receptor.
Sarcomas are a heterogeneous group of tumors. In the last 20 years, the finding of specific acquired chromosomal alterations in sarcomas has helped, in many cases, to understand their underlying genetic basis. Each of the various recurrent chromosomal translocations in sarcomas ultimately results in the creation of a novel chimeric or fusion gene. With the discovery of these new chromosomal alterations in sarcomas, and subsequently of the fusion genes, it was believed that remarkable progress would be possible in the treatment approach for these tumors. Certainly, our knowledge regarding different entities in sarcoma has considerably increased over the last two decades, and this information among others has helped to classify sarcomas more precisely and to identify new subgroups. However, whether this information has also translated into a better treatment approach is questionable.
In children, rhabdomyosarcoma (RMS) is the most common malignant soft-tissue tumor. Although combined treatment modalities including surgery, chemotherapy and/or radiotherapy have increased overall survival in many RMSs and other groups of soft-tissue sarcomas, conventional treatment has not yet resolved the problem of metastatic disease and many situations of disease relapse.
Furthermore, another important concern is the side effects of conventional chemotherapy and especially radiotherapy. Since most current therapies are not specifically targeted to tumor cells, a variety of normal cells in the body may also be affected by these therapeutic modalities, resulting in undesired and long-term side effects in the developing child.
Optimization of chemotherapy has certainly brought some improvement but it is unlikely to deliver a real breakthrough in the therapeutic approach. Combination therapies including vincristine, dactinomycin and cyclophosphamide or ifosfamide can be considered as the backbone of current treatment protocols. Other antineoplastic agents including doxorubicin, cisplatin, carboplatin, etoposide, topotecan, irinotecan, melphalan and methotrexate are active against RMS and have been combined in various combinations with backbone drugs. The overall survival rate in non-metastatic RMS has reached approximately 70%. However, substantial differences can be observed according to tumor site and clinical stage, ranging from approximately 60%, for example, in RMS of the limbs, to 90% or higher in orbital RMS or in genitourinary and nonbladder prostate RMS, respectively Citation[1,2]. Furthermore, histological subtype and tumor size as well as a patient’s age can significantly influence disease outcome.
In some of the reported treatment results, substantial differences between event-free and overall survival reaching up to 30%, for example, in orbital RMS, were observed Citation[2]. This reflects a philosophical attitude of treatment approaches for local disease control in different collaborative groups in the USA and Europe (the Intergroup Rhabdomyosarcoma Study Group [IRS], the International Society of Paediatric Oncology [SIOP] and the Cooperative Groups in Germany and Italy) Citation[3]. There is a continuous debate focusing on the method and timing of local therapy. This mainly includes the application and amount of radiotherapy and its long-term side effects for disease control, a treatment modality that is certainly necessary for many of the patients but not for all. However, it also indicates that for some patients, a successful re-treatment after relapse is possible. Beside the application of conventional radiotherapy, the use of brachytherapy or proton therapy has further increased the armamentarium of tools for local tumor control. Yet failure of local control remains a major cause of relapse in RMS. Whether a prolonged treatment approach is really superior to a standard treatment length is currently being tested in some randomized trials. In contrast to localized RMS, metastatic RMS is still a real challenge for the clinicians as the cure rate is very poor in spite of the initial response to chemotherapy. In more favorable risk groups, metastatic patients (aged <10 years without bone or bone marrow involvement) can reach survival rates of up to 62%, whereas in unfavorable subgroups (aged >10 years with presence of bone or bone marrow metastasis), a survival rate of 10% may not even achieved Citation[4,5]. High-dose chemotherapy followed by hematopoietic stem-cell rescue has not yet been proven to be beneficial in such situations. Therefore, improved and more specific (targeted) treatment approaches are urgently needed.
An impressive body of new biological information has been gathered during the last decades through modern cancer research. However, so far the majority of these insights could not be incorporated into more conventional treatment strategies as described above and the success of targeted therapies is limited to few, although striking, examples. The two main problems that emerged during early-phase clinical studies are selection of the right molecular targets including build-up of early resistance, and selection of the patients that would benefit from targeted therapeutic approaches. We will discuss these main issues in the context of childhood RMS in the following paragraphs.
Several molecular targets for RMS have been defined in the past, whereby the focus mainly concentrated on translocation-positive RMSs (tposRMS), which comprise approximately 80% of tumors with alveolar histology. Historically, RMS has been divided into two main subgroups based on histological criteria, namely, embryonal and alveolar RMS. However, molecular characterization of several hundred tumor biopsies by different laboratories has led to the conclusion that tumors bearing the characteristic PAX–FKHR (also called ‘PAX–FOXO1A’) translocation products are clearly distinct from all RMS tumors without translocation Citation[6], irrespective of histology. In agreement with these molecular data, one might also speculate that these two RMS subtypes have a different cell of origin and therefore represent two different diseases, although convincing experimental data supporting this conclusion are still lacking (for a more detailed discussion see Citation[7]). According to these findings, we therefore also discriminate between tposRMS and translocation-negative RMSs (tnegRMS) for this discussion.
The identification of molecular targets for cancer therapy of a given tumor type requires a large body of preclinical experiments to demonstrate addiction of tumor cells to one or more specific molecular pathways. Alternatively, and since cancer is recognized largely as a genetic disease, genetic analysis of tumors might reveal clear hints regarding possible targets. In this respect, again different genetics underlie tnegRMS and tposRMS . Most strikingly of course, this is illustrated by the translocation products themselves, which are widely thought to be the transformation initiating event. Therefore, these tumor-specific fusion proteins might represent ideal therapeutic targets, although they are likely to require additional cooperating events. This notion was also experimentally validated by using different knock-down strategies, which showed, both in vitro as well as in vivo, that tposRMS cells require the expression of the fusion protein for tumor cell survival and therefore tumor maintenance Citation[8–10]. Of course, a range of other very promising therapeutic targets/pathways are being evaluated for tposRMS by several investigators including the IGF, FGF and MET pathways (see ). Strikingly, components of these pathways, such as their receptor tyrosine kinases, are direct target genes of the fusion proteins. This further underscores the importance of the chromosomal translocation products. Therefore, a reduction in transcriptional activity of PAX–FKHR might lead to the development of effective therapeutic strategies, affecting multiple signaling pathways at the same time. As transcription factors cannot be targeted directly by small molecular drugs, interference with activity at different levels of regulation can be envisaged. These include, for example, post-translational modifications, protein–protein interactions, protein stability or chromatin modeling at target gene promoters . Combining strategies that interfere with transcriptional activation and at the same time restore expression of repressed genes might be especially promising Citation[11]. Indeed, current evidence suggests that for most targeted therapies, combinations of drugs might be needed to achieve improvements in survival rates and to prevent the rapid appearance of resistant clones. These combinations include the addition of targeted drugs to existing therapies, or combinations of targeted drugs given as a window therapy. Since the number of patients with pediatric sarcomas who are eligible for early clinical trials is very limited, such combinations need to be evaluated in preclinical models. Establishing primary ‘tumorgrafts’ in mice might be a first step towards this goal.
Today, only very few clinical trials incorporating new agents into RMS treatment have been conducted, the majority of which recruit patients over a broad spectrum of pediatric sarcomas to increase the number of eligible patients. In doing so, any clinical benefit in a subgroup of patients might be masked by unresponsive patient groups. Therefore, it will be critical for future translational studies to select patients according to the targeted agent(s) to be tested. In the absence of specific biomarkers, this might be accomplished most easily through genetic analysis, for example, by determination of the fusion gene or mutational status if possible.
Multidisciplinary management of childhood sarcomas incorporating different medical subspecialties has improved patient prognosis during the last decades. However, further improvements might hopefully come from the incorporation of newly developed targeted agents into treatment strategies. It is specifically in this field where clinicians and scientists have to fuse their knowledge and learn how to work together successfully and efficiently in treatment management. Ultimately, by expanding the multidisciplinary approach, we hope to apply therapies that are sufficiently specific and effective and, thus, may truly be ‘magic bullets’.
Table 1. Selection of genetic alterations representing potential drug targets in rhabdomyosarcoma.
Financial & competing interests disclosure
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
References
- Dantonello TM, Int-Veen C, Harms D et al. Cooperative trial CWS-91 for localized soft tissue sarcoma in children, adolescents, and young adults. J. Clin. Oncol.27(9), 1446–1455 (2009).
- Stevens MC, Rey A, Bouvet N et al. Treatment of nonmetastatic rhabdomyosarcoma in childhood and adolescence: third study of the International Society of Paediatric Oncology – SIOP Malignant Mesenchymal Tumor 89. J. Clin. Oncol.23(12), 2618–2628 (2005).
- Stevens MC. Philosophy of treatment and the role of chemotherapy in paediatric soft tissue sarcomas. Eur. J. Cancer Suppl.1, 237–247 (2003).
- Carli M, Colombatti R, Oberlin O et al. European intergroup studies (MMT4–89 and MMT4–91) on childhood metastatic rhabdomyosarcoma: final results and analysis of prognostic factors. J. Clin. Oncol.22(23), 4787–4794 (2004).
- McDowell HP, Foot AB, Ellershaw C et al. Outcomes in paediatric metastatic rhabdomyosarcoma: results of the International Society of Paediatric Oncology (SIOP) study MMT-98. Eur. J. Cancer46(9), 1588–1595 (2010).
- Williamson D, Missiaglia E, de Reynies A et al. Fusion gene-negative alveolar rhabdomyosarcoma is clinically and molecularly indistinguishable from embryonal rhabdomyosarcoma. J. Clin. Oncol.28(13), 2151–2158 (2010).
- Charytonowicz E, Cordon-Cardo C, Matushansky I, Ziman M. Alveolar rhabdomyosarcoma: is the cell of origin a mesenchymal stem cell? Cancer Lett.279(2), 126–136 (2009).
- Ayyanathan K, Fredericks WJ, Berking C et al. Hormone-dependent tumor regression in vivo by an inducible transcriptional repressor directed at the PAX3–FKHR oncogene. Cancer Res.60(20), 5803–5814 (2000).
- Bernasconi M, Remppis A, Fredericks WJ, Rauscher FJ 3rd, Schafer BW. Induction of apoptosis in rhabdomyosarcoma cells through down-regulation of PAX proteins. Proc. Natl Acad. Sci. USA93(23), 13164–13169 (1996).
- Kikuchi K, Tsuchiya K, Otabe O et al. Effects of PAX3-FKHR on malignant phenotypes in alveolar rhabdomyosarcoma. Biochem. Biophys. Res. Commun.365(3), 568–574 (2008).
- Hecker RM, Amstutz RA, Wachtel M et al. p21 downregulation is an important component of PAX3/FKHR oncogenicity and its reactivation by HDAC inhibitors enhances combination treatment. Oncogene29(27), 3942–3952 (2010).
- Denayer E, Devriendt K, de Ravel T et al. Tumor spectrum in children with Noonan syndrome and SOS1 or RAF1 mutations. Genes Chromosomes Cancer49(3), 242–252 (2009).
- Martinelli S, McDowell HP, Vigne SD et al. RAS signaling dysregulation in human embryonal rhabdomyosarcoma. Genes Chromosomes Cancer48(11), 975–982 (2009).
- Zibat A, Uhmann A, Nitzki F et al. Time-point and dosage of gene inactivation determine the tumor spectrum in conditional Ptch knockouts. Carcinogenesis30(6), 918–926 (2009).
- Anderson J, Gordon A, McManus A, Shipley J, Pritchard-Jones K. Disruption of imprinted genes at chromosome region 11p15.5 in paediatric rhabdomyosarcoma. Neoplasia1(4), 340–348 (1999).
- Cao L, Yu Y, Darko I et al. Addiction to elevated insulin-like growth factor I receptor and initial modulation of the AKT pathway define the responsiveness of rhabdomyosarcoma to the targeting antibody. Cancer Res.68(19), 8039–8048 (2008).
- Wachtel M, Runge T, Leuschner I et al. Subtype and prognostic classification of rhabdomyosarcoma by immunohistochemistry. J. Clin. Oncol.24(5), 816–822 (2006).
- Ganti R, Skapek SX, Zhang J et al. Expression and genomic status of EGFR and ErbB-2 in alveolar and embryonal rhabdomyosarcoma. Mod. Pathol.19(9), 1213–1220 (2006).
- Bennicelli JL, Barr FG. Chromosomal translocations and sarcomas. Curr. Opin. Oncol.14(4), 412–419 (2002).
- Mercado GE, Xia SJ, Zhang C et al. Identification of PAX3–FKHR-regulated genes differentially expressed between alveolar and embryonal rhabdomyosarcoma: focus on MYCN as a biologically relevant target. Genes Chromosomes Cancer47(6), 510–520 (2008).
- Taylor JG, Cheuk AT, Tsang PS et al. Identification of FGFR4-activating mutations in human rhabdomyosarcomas that promote metastasis in xenotransplanted models. J. Clin. Invest.119(11), 3395–3407 (2009).
- Schaaf G, Hamdi M, Zwijnenburg D et al. Silencing of SPRY1 triggers complete regression of rhabdomyosarcoma tumors carrying a mutated RAS gene. Cancer Res.70(2), 762–771 (2010).
- Oesch S, Walter D, Wachtel M et al. Cannabinoid receptor 1 is a potential drug target for treatment of translocation-positive rhabdomyosarcoma. Mol. Cancer Ther.8(7), 1838–1845 (2009).
- Davicioni E, Finckenstein FG, Shahbazian V et al. Identification of a PAX–FKHR gene expression signature that defines molecular classes and determines the prognosis of alveolar rhabdomyosarcomas. Cancer Res.66(14), 6936–6946 (2006).
- Taulli R, Scuoppo C, Bersani F et al. Validation of met as a therapeutic target in alveolar and embryonal rhabdomyosarcoma. Cancer Res.66(9), 4742–4749 (2006).