Abstract
Agonists of 5-hydroxytryptamine (5-HT; serotonin) receptors promote loss of excessive body fat (adiposity) and improve metabolic parameters associated with adiposity-induced adipose tissue dysfunction (adiposopathy or ‘sick fat’). By improving adipose tissue pathogenic endocrine and immune responses in overweight patients, 5-HT receptor agonists may improve metabolic disease. Lorcaserin (APD-356) is a selective 5-HT2c receptor agonist that promotes weight loss. Probably owing to its selectivity for the 5-HT2c receptor, clinical trial evidence supports that lorcaserin does not adversely affect heart valves or pulmonary artery pressure. This review examines: the mechanisms by which serotonergic pathways improve adiposity and adiposopathy; historical data and perspective regarding the efficacy and safety of prior 5-HT agonists; speculation regarding future paradigms in treating adiposopathy; and why lorcaserin may prove to be a safe and generally well-tolerated agent that not only improves the weight of patients, but also improves the health of patients.
Increased CNS serotonin, leptin and/or insulin activity increases anorexigenic POMC (which is cleaved to form melanocortins such as MSH) and CART expression, and decreases orexigenic expression of NPY and AgRP. Alterations in these peptidergic appetitive effectors contribute to ‘second order’ signaling through arcuate nucleus neurons projection to other hypothalamic regions. The net result of increased CNS serotonergic activity is increased anorexigenic and increased catabolic effects, with decreased orexigenic and decreased anabolic effects. This figure is greatly simplified, and does not show all the interconnected signaling pathways between these CNS factors.
AgRP: Agouti-related peptide; BDNF: Brain-derived neurotrophic factor; CART: Cocaine amphetamine-regulated transcript; CB1: Cannabinoid 1 receptor; CRH: Corticotropin-releasing hormone; MC3R: Melanocortin 3 receptor; MC4R: Melanocortin 4 receptor; MCH: Melanin concentrating hormone; MSH: Melanocyte stimulating hormone; NPY: Neuropeptide Y; POMC: Pro-opiomelanocortin; TRH: Thyroid releasing hormone.
![Figure 1. Simplified relationship between serotonin (and other selected CNS factors) on anorexigenic and orexigenic peptidergic appetitive effectors within the hypothalamus Citation[36,112].Increased CNS serotonin, leptin and/or insulin activity increases anorexigenic POMC (which is cleaved to form melanocortins such as MSH) and CART expression, and decreases orexigenic expression of NPY and AgRP. Alterations in these peptidergic appetitive effectors contribute to ‘second order’ signaling through arcuate nucleus neurons projection to other hypothalamic regions. The net result of increased CNS serotonergic activity is increased anorexigenic and increased catabolic effects, with decreased orexigenic and decreased anabolic effects. This figure is greatly simplified, and does not show all the interconnected signaling pathways between these CNS factors.AgRP: Agouti-related peptide; BDNF: Brain-derived neurotrophic factor; CART: Cocaine amphetamine-regulated transcript; CB1: Cannabinoid 1 receptor; CRH: Corticotropin-releasing hormone; MC3R: Melanocortin 3 receptor; MC4R: Melanocortin 4 receptor; MCH: Melanin concentrating hormone; MSH: Melanocyte stimulating hormone; NPY: Neuropeptide Y; POMC: Pro-opiomelanocortin; TRH: Thyroid releasing hormone.](/cms/asset/469b8346-307f-40c3-9cbf-7677a414489b/ierk_a_11210497_f0001_b.jpg)
*‘Lorcaserin’ is listed as an illustrative example of an anti-obesity agent that alone, or possibly in combination with other anti-obesity agents, may improve adiposopathy. The intent is to improve the otherwise dysfunctional adipose tissue in overweight patient, and thus correct the pathogenic endocrine and immune derangements that lead to metabolic disease.
Agonism of the 5-HT receptor promotes weight loss, which may improve fat mass-related morbidities in overweight patients Citation[1]. Weight loss, such as through serotinergic agents, may also improve pathogenic adipose tissue endocrine and immune responses in overweight patients, which, in turn, may also improve adiposity-related metabolic disease. Adiposopathy (‘sick fat’) is a term that describes pathogenic adipose tissue (Box 1)Citation[2]. Anatomically, adiposopathy is manifest by adipocyte hypertrophy, adipose tissue accumulation (particularly visceral adiposity), adipose tissue growth beyond its vascular supply (potentially resulting in hypoxia), limitations of adipose tissue growth (e.g., impaired extracellular matrix dissolution and remodeling), and ‘lipotoxic’ fat (free fatty acid and triglyceride) deposition in ectopic peripheral organs such as liver, muscle and pancreas Citation[2–8]. These adipocyte and adipose tissue anatomic derangements are promoted by positive caloric balance and sedentary lifestyle in genetically and environmentally susceptible patients Citation[3]. Pathophysiologically, these anatomic abnormalities lead to adverse adipose tissue endocrine and immune responses that contribute to metabolic disease Citation[9]. Adiposopathy represents a unified, pathophysiologic process that is a root cause of metabolic diseases such as Type 2 diabetes mellitus, high blood pressure and dyslipidemia Citation[10]. However, just as an increase in bodyweight may promote pathogenic adipose tissue responses Citation[3,11], a reduction in body fat in overweight patients with metabolic diseases can often improve pathogenic adipose tissue responses, and thus improve metabolic disease Citation[12,13].
Clinical overview
Obesity is an epidemic. In the USA, over 30% of adults are obese, with over 65% being overweight (BMI ≥ 25 kg/m2) or obese (BMI ≥ 30 kg/m2) Citation[201]. Globally, more than 1 billion adults are overweight, with at least 300 million being obese Citation[202]. Given the number of lives affected, obesity may represent the greatest epidemic in human existence. An increase in bodyweight is directly associated with the most common metabolic diseases encountered in the clinical practice of medicine, and increases the risk of Type 2 diabetes mellitus, high blood pressure and dyslipidemia Citation[11,14].
Recognizing the contribution of adiposity to metabolic disease is sometimes obscured, at least in part, due to apparent ‘obesity paradoxes’. While excessive bodyweight is generally considered unhealthy, substantial data suggest that many modestly overweight patients experiencing atherosclerotic coronary heart disease (CHD) events, or undergoing CHD procedures, have better health outcomes than those who are not overweight Citation[15,16]. Another apparent paradox is that individuals who are modestly overweight may live longer than many of those who weigh less Citation[17]. Yet another obesity paradox is that not all overweight patients have metabolic disease, and not all patients with metabolic disease are overweight Citation[11,18]. These apparent ‘paradoxical’ findings may be less paradoxical upon recognizing that adiposity leading to metabolic disease may be mitigated if weight gain occurs without pathogenic endocrine or immune adipose tissue responses, or at least not beyond what can be appropriately managed (metabolically) by other body tissues Citation[11]. Conversely, some individuals (and some genetically predisposed populations) who undergo only modest fat weight gain may have pathogenic adipose tissue responses that lead to metabolic disease Citation[6,3]. Thus, adiposity’s contribution to metabolic disease is significantly dependent upon how the fat is gained (e.g., adipogenesis), where the fat is gained (e.g., visceral versus subcutaneous adipose tissue), the effect upon adipose tissue functionality (e.g., endocrine and immune responses, as well as net free fatty acid release), and interactions (e.g., ‘cross talk’) with other body tissues (Box 1)Citation[3,11]. From a treatment standpoint, the effectiveness of any anti-obesity therapy must take into account the degree to which it not only reduces bodyweight, but also improves adipose tissue function and patient health.
Perhaps the most cost-effective way for the individual obese patient to lose weight is through improved nutrition and increased physical activity. However, irrespective of numerous existing public health initiatives and commercial programs, interventions to promote clinically meaningful and sustained weight loss among the general population have proven to be mostly unsuccessful Citation[19]. The ongoing obesity epidemic continues unabated. Another treatment option includes bariatric surgery, which is becoming a more frequent and widespread intervention that not only effectively treats obesity, but also effectively (and sometimes dramatically) improves adiposopathy-associated metabolic diseases Citation[8,20].
Surveys suggest that approximately 30% of men and 40% of women report being on dietary and/or prescription/nonprescription therapy for the purposes of losing weight; many do so for reasons other than to improve their health Citation[21]. For those with adverse health consequences, such as adiposity-related mass disorders or adiposopathy-related metabolic disease, anti-obesity drug therapies are indicated as an adjunct to appropriate nutrition and physical activity for the purpose of weight reduction. These agents are also often effective in improving adipose tissue endocrine and immune responses Citation[13], as well as improving metabolic disease Citation[12,22]. Unfortunately, many of the existing anti-obesity agents have adverse tolerability or safety effects that limit their use Citation[13]. To help address these limitations on the more widespread use of anti-obesity agents, investigational combination anti-obesity agents are in late-stage development. Examples include Contrave®, which is a fixed-dose combination of sustained-release bupropion 360 mg (a nonselective dopamine and norepinephrine-reuptake inhibitor) and sustained-release naltrexone 16–32 mg (an opioid receptor antagonist) Citation[13,23], and Empatic™, which is a fixed-dose combination of sustained-release zonisamide 120–360 mg and sustained-release bupropion 360 mg Citation[24]. Qnexa™ is also an investigational anti-obesity agent that is a combination agent of phentermine 3.75–15 mg and controlled-release topiramate 23–92 mg Citation[13,25].
It is noteworthy that of the five weight loss drugs that currently have US FDA approval for weight loss, only two are approved for long-term use (up to 1 year): orlistat and sibutramine Citation[203]. The lack of a greater number of long-term anti-obesity drug treatment options is significantly (and in some ways, uniquely) due to a dismal history of high-profile development failures and post-marketing withdrawals as the result of safety and tolerability concerns Citation[26–28]. Another impediment to the success of anti-obesity pharmaceutical therapy is that the weight loss achieved with these agents is often variable, and when achieved, often modest. This is problematic because of the frequent perception disconnect between the degree of weight loss meeting patient expectations, and that which might be clinically meaningful in improving patient health. Strictly from a health standpoint, even a 5% weight loss may improve the adipose tissue pathogenic responses characteristic of adiposopathy Citation[29]. Thus, even a mild-to-modest weight loss of 5–10% in overweight patients may help prevent the onset of, or provide clinically meaningful therapeutic benefits in the treatment of, Type 2 diabetes mellitus, high blood pressure and/or dyslipidemia Citation[30,31]. The practical implication is that the focus of any weight loss investigational agent should have the following caveat:
“The development of any effective anti-obestiy agent must not only reduce fat mass (adiposity), but must also correct fat dysfunction (adiposopathy) in order to maximize metabolic healthCitation[32].’”
Serotonin pathways
Appetite is derived from the Latin word appetitus, meaning ‘desire.’ The hypothalamus is a major hub region of the brain that significantly controls desirous behavior, such as fighting, fleeing, mating and feeding. Appetite and behaviors related to feeding are dependent upon a complex and redundant system of endocrine receptors, transporters and neurocircuits that integrate signaling from the periphery (e.g., gastrointestinal system, adipose tissue, liver, muscle and pancreas) and within the CNS Citation[33–35]. Serotonin is a biogenic amine neurotransmitter whose decrease in CNS bioavailability promotes hyperphagia and weight gain, and whose increase in bioavailability promotes decreased food intake and weight loss. Increased CNS serotonergic activity through increased G-protein-coupled 5-HT 2c receptor agonism increases anorexigenic pro-opioidmelanocortin (POMC) production in the arcuate nucleus area in the hypothalamus. Increased CNS serotonergic activity through increased G-protein-coupled 5-HT 1b receptor agonism decreases orexigenic neuropeptide Y (NPY) production, which is also in the arcuate nucleus area in the hypothalamus Citation[36]. The net result of increased CNS serotoninergic activity is an increase in anorexigenic and catabolic effects, a decrease in orexigenic and anabolic effects, and therefore, weight loss . Animal studies suggest that 5-HT2c receptor null mice have depressed resting metabolic rate, as well as hyperphagia and disrupted satiety Citation[37]. In humans, some studies suggest that 5-HT receptor agonism may increase resting metabolic rate and thermogenesis Citation[38,39]. Other studies do not support that 5-HT agonism increases resting metabolic rate Citation[40–42]. Thus, while 5-HT receptor agonism might possibly potentiate energy expenditure with physical activity, or perhaps even increase the thermal effect of food Citation[43], the greatest effect of 5-HT agonism is probably due to decreased appetite and diminished food intake Citation[44,45].
The effects of serotonin are mediated through at least 14 subtypes of the serotonin receptor family Citation[46–48]. Applicable to this discussion are the G-coupled 5-HT2 receptor subfamily, which contains 5-HT2c, 5-HT2a and 5-HT2b receptors. The 5-HT2c receptors are located almost exclusively in the CNS, particularly in the choroid plexus, as well as limbic system structures that help control behaviors such as the hypothalamus and thalamus Citation[47,49]. 5-HT2c receptors are involved in the brain process involving caloric balance and are a target for anti-obesity drug therapies . They may also be associated with CNS processes involving mood and cognition, and 5-HT2c receptors are also potential targets for epilepsy, obsessive–compulsive disorders, Parkinson’s diseases, schizophrenia, depression, anxiety, sleep disorders and drug abuse Citation[48]. 5-HT2a receptors are likewise located in the CNS, mainly in the cortex and hippocampus Citation[50], and are targets for various psychiatric and sleep therapeutic agents. 5-HT2a receptors may also be located on cardiac vessels and heart valves Citation[51,52]. 5-HT2b receptors are found on cardiac vessels Citation[51] and heart cells, such as cardiac fibroblasts and cardiac valves. An increase in serotonin stimulation of 5-HT2b receptors, as occurs with carcinoid syndrome, may cause cardiomyopathy and endocardial fibrosis of heart valves Citation[53]. 5-HT2b receptor antagonists are being evaluated as a treatment for heart failure Citation[54,55].
Lorcaserin
Lorcaserin is a small-molecule, selective 5-HT2c receptor agonist that is being developed as a weight-loss drug therapy. Lorcaserin (formerly known as APD-356) is chemically described as [1R]-8-chloro-2,3,4,5-tetrahydro-1-methyl-1H-3-benzazepine Citation[49]. Lorcaserin is a selective 5-HT2c receptor agonist with 15- and 100-fold functional selectivity versus 5-HT2A and 5-HT2B receptors, respectively Citation[48,49]. Increased CNS serotonin activity through stimulation of the 5-HT2c receptor modulates caloric balance by activating the POMC system, and thus affects downstream pathways, promoting catabolism and inhibiting anabolism Citation[56,57].
In preclinical studies involving animals (rat), locaserin underwent rapid gastrointestinal absorption with maximum blood levels in approximately 0.5 h and maximum brain exposure in approximately 1 h. The elimination half-life from both blood and brain was approximately 4–6 h. The major circulating metabolite did not appreciably bind to 5-HT2 receptors Citation[48]. In humans, the mean first day (± standard deviation) Cmax for lorcaserin for 10 and 15 mg once a day, and 10 mg twice a day was approximately 157 ± 62, 230 ± 96 and 158 ± 56 nmol/l, respectively. In Phase I studies of a lorcaserin 10-mg dose, steady state was achieved by day 4, with Cmax values approximately 30% higher than on day 1 Citation[49]. In single-dose studies of oral administration to healthy volunteers, pharmacokinetic analysis Citation[58] of lorcaserin 10, 20 and 40 mg stopped at 40 mg per day, due to disorientation and limb unawareness in a single woman subject at the 40 mg dose. A high-fat meal resulted in a 2-h delay in Tmax of lorcaserin, but otherwise, the pharmacokinetic properties were unaffected. The Tmax was approximately 2 h for all doses. Radiolabeled lorcaserin was mainly metabolized to a sulfamate metabolite, among other metabolites. This sulfamate metabolite represented 38% of blood radioactivity while the parent drug (lorcaserin) was 12% of blood radioactivity. The major route of elimination of radiolabel lorcaserin and its metabolites was the urine (92%), and the major urine metabolite was the lorcaserin N-carbamoyl glucuronide. Lorcaserin’s half-life was approximately 10–11 h.
Clinical efficacy of 5-HT agonists
Historical perspective of 5-HT receptor agonist efficacy
Increasing CNS 5-HT2c activity as a mechanism to promote weight loss is neither new, nor novel. Prior to lorcaserin, various 5-HT receptor agonists validated 5-HT receptors as an effective target to treat obesity. Fenfluramine was an amphetamine derivative (without psychostimulant activities) developed in the 1960’s that nonselectively stimulated 5-HT receptors, including 5-HT2c. It was approved as an anti-obesity agent in 1973 Citation[26,36]. One of several earlier, yet illustrative, studies of fenfluramine evaluated ‘refractory’ obese women. ‘Obesity’ was defined as at least 20% above ‘standard weight’ as determined by the USA Medico Actuarial Investigation data of 1912. The study report described participants as ‘mostly middle-aged housewives’ and with no known cardiac disease or edema Citation[59]. Of the 60 women who began the 12-week trial, 50 completed (25 in the fenfluramine group and 25 in the ‘dummy’ control group). The fenfluramine group lost 9.3 lbs, while the control group gained 0.4 lbs. The conclusion was:
“Thus, fenfluramine, although expensive, appears to be a more effective appetite suppressant than other anorectic drugs that we have so far tested. However, a mean weight loss of only 9.3 lbs (4220 g) in 12 weeks is still not very substantial.”
A review of multiple studies over the ensuing decades found that, compared with placebo: fenfluramine reduced bodyweight in obese patients by approximately 3 kg (∼6–7 lbs); more patients in the fenfluramine-treated group dropped out of studies due to adverse drug effects (9 vs 2% for placebo); and fewer patients dropped from studies due to perceived lack of weight loss (3 vs 6% for placebo) Citation[60]. Importantly, through trials conducted in the 1970s–1990s, fenfluramine improved glucose levels in patients with Type 2 diabetes mellitus Citation[61], and improved blood pressure Citation[62] and lipid levels Citation[61,63]. This helped validate that even modest weight loss through 5-HT receptor agonism had the potential to not only improve the weight of patients, but also improve the metabolic health of patients.
As levofenfluramine was thought to have potential adverse effects while providing no additional efficacy, and because fenfluramine contained both levofenfluramine and dexfenfluramine, dexfenfluramine (the active isomer) underwent clinical evaluation. The intent was to improve upon the efficacy, safety and tolerability of this class of agents through development of the dextrostereoisomer component of fenfluramine, which would presumably represent an anti-obesity agent that was a ‘pure’ serotonin receptor agonist, devoid of additional adverse antidopaminergic or sympathomimetic effects Citation[64]. In 1996, dexfenfluramine became the first anti-obesity drug approved in the USA for long-term clinical use (then defined as over 3 months) Citation[47]. (Sibutramine was approved in the USA in 1997; orlistat was approved in the USA in 1999) Citation[26].
Despite the promise of both fenfluramine and dexfenfluramine, concerns persisted regarding the modest degree of weight loss, and the maintenance of clinically meaningful weight loss beyond 3 months Citation[65]. A lack of more robust weight loss with these agents as monotherapy prompted the increased ‘off label’ use of fenfluramine combined with phentermine (a sympathomimetic adrenergic agent with limited dependence potential). This combination became popularly known as ‘fen–phen’ or ‘phen–fen.’ Such an approach and strategy had scientific backing, largely based upon a preliminary 24-week study published in 1984 Citation[66], and especially after a series of articles published in 1992 regarding a clinical trial of 121 overweight patients, defined as 130–180% of their ideal bodyweight (IBW) as determined by the 1983 Metropolitan Life tables. In this study, at 34 weeks, fen–phen-treated subjects lost approximately 14 kg (∼31 lbs) compared with approximately 5 kg in the placebo group (∼10 lbs). Other than dry mouth, this combination treatment was generally well tolerated. Subsequent publications focused on analyses at weeks 34–104 Citation[67], 104–156 Citation[68], 156–190 Citation[69] and 190–210 Citation[70], and supported a reasonable degree of continued efficacy of the combination of fenfluramine and phentermine in treating obesity and metabolic complications for as long as 4 years Citation[71,72]. However, less than a third of participants completed the study, and most regained weight during its latter stages Citation[26]. Some characterized this ‘single study’ as ‘fueling the phen–fen craze’Citation[26]. In 1997, just a little over a year after approval of dexfenfluramine, and over two decades after approval of fenfluramine, both dexfenfluramine and fenfluramine were withdrawn from the market due to an increased risk of valvular heart disease Citation[13] – a cardiac toxicity subsequently thought due to the nonselective stimulation of cardiac 5-HT2b receptors (see discussion on safety, later).
Given that the established efficacy of these 5-HT receptor agonists was thought to be largely due to stimulation of the 5-HT2c receptor, and given that the cardiac valvular toxicity was thought to be likely due to stimulation of the 5-HT2b receptor, efforts were then directed towards the discovery and development of anti-obesity agents that were selective for the 5-HT2c receptor Citation[44]. An example was APD-356, which was subsequently named lorcaserin Citation[73,48].
Lorcaserin efficacy: Phase II clinical trial data
In 2005, 8 years after withdrawal of fenfluramine and dexfluramine, recruitment began on a sentential Phase II lorcaserin clinical trial. This was a 12-week, randomized, double-blind, placebo-controlled, parallel-arm study of 469 men and women (mean: ∼13% men; ∼87% women) 18–65 years of age (mean: ∼42 years) with a BMI of 30–45 kg/m2 (mean: ∼36 kg/m2). Participants did not have significant dyslipidemia or hypertension, did not have diabetes mellitus, and could remain on stable antihypertensive or lipid-altering drug therapy. Participants received instructions to maintain their ‘usual’ nutritional intake. Baseline mean waist circumference was approximately 108 cm (one out of the five diagnostic components of the metabolic syndrome is increased waist circumference, which is defined as over 102 cm [>40 inches] for men, and over 88 cm [>35 inches] for women Citation[74]). This study had a high representation of minority populations, with approximately 52% described as white, approximately 29% described as black, and approximately 18% described as being Hispanic. Among completers, the treatment groups of placebo, lorcaserin 10 mg once per day, lorcaserin 15 mg once per day, and lorcaserin 10 mg twice a day produced weight loss of 0.3, 1.8, 2.6 and 3.6 kg (p < 0.001 vs placebo for each group). The proportions of completers achieving 5% or more of initial bodyweight were 2.3, 12.8, 19.5 and 31.2%, respectively. Lorcaserin-associated weight loss also produced significant reductions waist (and hip) circumference at the 10 mg twice a day dose.
Mean baseline systolic and diastolic blood pressure was approximately 121/78 mmHg. Lorcaserin did not produce significant changes in blood pressure. Mean baseline total cholesterol (TC) was approximately 195 mg/dl (∼5 mmol/l). Lorcaserin 10 mg twice a day lowered TC by approximately 7 mg/dl (-0.18 mmol/l; p < 0.01). Mean baseline triglyceride (TG) level was approximately 134 mg/dl (∼1.5 mmol/l). Lorcaserin 10 mg twice a day lowered TG levels approximately 18 mg/dl (-0.2 mmol/l; p = not significant [NS]); Mean baseline LDL-cholesterol level was approximately 113 mg/dl (∼2.9 mmol/l). Lorcaserin 10 mg twice a day lowered LDL-cholesterol by approximately 4 mg/dl (-0.1 mmol/l; p = NS). Mean baseline HDL-cholesterol was 59 mg/dl (∼1.5 mmol/l). Lorcaserin 10 mg twice a day lowered HDL-cholesterol by approximately 4 mg/dl (∼0.1 mmol/l; p < 0.05). This mild reduction in HDL-cholesterol was not unexpected in this short trial, and consistent with a prior meta-analysis of 70 clinical trials, which concluded that while HDL-cholesterol levels often rise after weight loss stabilization, HDL-cholesterol levels typically fall during active weight loss Citation[75]. Baseline mean fasting glucose level was approximately 92 mg/dl (∼5.1 mmol/l). Lorcaserin 10 mg twice a day significantly lowered glucose levels approximately 4 mg/dl (-0.2 mmol/l; p < 0.05). Thus, this 12-week trial supported lorcaserin as efficacious in producing progressive, dose-dependent, significant weight loss. Lorcaserin at the 10-mg twice daily dose also improved several metabolic parameters compared with placebo, even without implementation of concomitant nutritional or physical activity interventions. In total, this Phase II trial supported lorcaserin as not only improving the weight of patients, but also improving the metabolic health of patients.
Mechanisms by which 5-HT agonists may improve adiposopathy & metabolic disease
As with other interventions, the mechanisms by which 5-HT2 agonists (such as lorcaserin) improve metabolic efficacy parameters are likely multifactorial ; . The primary effects of most weight loss agents that improve metabolic disease are a reduction in body fat (through reduction in appetite), and subsequent improvement in adipose tissue anatomy accompanied by improvements in adipose tissue endocrine and immune responses. Many metabolic derangements associated with increased bodyweight can be attributable to adiposopathy, or ‘sick fat,’ which may be described as having six main components, or ‘six faces’ Citation[12]. While many of the specific effects of lorcaserin on these components have yet to be studied or reported, the following is a brief consideration of what is known about serotoninergic effects that may affect adipose tissue’s pathogenic potential.
Adipogenesis
Adipogenesis includes the recruitment, proliferation and differentiation of new fat cells. The capacity for adipogenesis is genetically and environmentally determined, and influenced by adipocyte and nonadipocyte processes Citation[3]. If adipocyte proliferation is impaired during positive caloric balance, then existing adipocytes may undergo excessive lipogenesis and hypertrophy. This may lead to pathogenic adipocyte and adipose tissue endocrine and immune dysfunction (adiposopathy or ‘sick fat’) that, in turn, contributes to metabolic disease. Similarly, if adipocyte differentiation is impaired during positive caloric balance, then this may also result in adipose tissue dysfunction Citation[6].
Peripheral 5-HT2 receptors (e.g., 5-HT2a and 5-HT2b) receptors may play a direct role in adipocyte differentiation Citation[204]. However, because 5-HT2c receptors are virtually exclusive to the CNS, then 5-HT2c receptor acting agents would not likely have direct effects upon adipogenesis. However, increased activity of 5-HT2c receptors may have indirect effects on adipogenesis and adipose tissue metabolism, as is outlined in . Despite these potential indirect effects, it is likely that the greatest effect of 5-HT2c upon adipogenesis is related to the weight loss that occurs with CNS signaling that promotes negative caloric balance. In other words, the most likely mechanism by which 5-HT2c may affect adipogenesis and adipogenic markers is through a reduction in caloric balance and reduction in adiposity.
But as with other weight loss interventions, markers for adipogenesis during active weight loss should be interpreted within the context of active weight loss or maintenance of weight loss Citation[8]. This is important because, during positive caloric balance, reduced or impaired adipogenesis contributes to pathogenic adiposopathic endocrine and immune responses that promote metabolic disease Citation[3,6]. By contrast, the decrease in adipogenic markers with negative caloric balance and weight loss Citation[8] does not necessarily mean that adipocytes or adipose tissue are becoming dysfunctional or ‘sick’. Instead, it is likely that adipogenesis is not physiologically appropriate (i.e., not needed) during negative caloric balance. Nonetheless, while adipogenic markers may decrease during negative caloric balance Citation[12], the capacity for subsequent adipogenesis may be increased. In fact, enhanced preadipocyte adipogenic and antiapoptotic protein expression, and increased adipogenic potential might help contribute to the high rate of weight regain among overweight patients who lose weight Citation[76]. It is therefore possible that one of the more important mechanisms by which weight loss improves or prevents metabolic disease is through an enhanced adipocyte capacity for unencumbered adipogenesis during future positive caloric balance, which is made even more favorable if positive caloric balance never recurs.
Pathogenic adipose tissue location
The pathogenic potential of adipose tissue is not only based upon how the fat is stored, but also where the fat is stored. While all adipose tissue stores may have pathogenic potential Citation[3,6], visceral adipose tissue is generally considered to be more pathogenic than abdominal subcutaneous adipose tissue, which, in turn, may be more pathogenic than peripheral subcutaneous adipose tissue Citation[3]. Visceral and other fat depots differ in genetic predisposition regarding metabolic activity, functionality, production of bioactive molecules, activity of various adipocyte receptors, and enzymatic processes involved with fat metabolism Citation[3]. Visceral adipose tissue also differs by its location, having circulatory drainage to the liver through the portal vessel system. Visceral adipose tissue has higher sensitivity to catecholamines, and lower sensitivity to insulin compared with subcutaneous, peripheral fat. Owing to physiologic differences such as these, visceral fat accumulation is a better predictor of metabolic disease than overall adiposity, and is likely a better treatment target to reduce metabolic disease than BMI alone Citation[77–81]. In consideration of both how and where fat is stored Citation[3], adipocyte hypertrophy, particularly in the visceral region, will often worsen adipocyte function resulting in increased free fatty acid release into the circulation Citation[4], and pathogenic endocrine and immune adipose tissue responses that contribute to metabolic disease Citation[3,5,18,82].
Most weight-loss interventions result in a decrease in total body fat, including both visceral and subcutaneous adipose tissue. While not a direct measure, waist circumference is an often used surrogate for visceral fat assessment. Through direct measures of body composition by whole-body, dual-energy, x-ray absorptiometry (DEXA), as well as indirect measures, dexfenfluramine reduces bodyweight, fat mass, fat-free mass, waist circumference and hip circumference Citation[83]. Similarly, lorcaserin reduces total bodyweight, BMI and waist circumference Citation[49]. While animal studies support the weight reduction with lorcaserin to be preferentially fat weight loss rather than lean weight loss Citation[48], the effects of lorcaserin on adipocyte size, adipocyte number, and more direct specific measures on visceral adiposity (DEXA) have yet to be reported.
Free fatty acid release
A net increase in free fatty acids occurs when adipocyte lipolysis exceeds lipogenesis, and is an important adipose tissue pathogenic response to adiposity and adiposopathy Citation[5]. Increased circulating free fatty acids are lipotoxic to other body organs such as liver, muscle and pancreas, resulting in insulin resistance, diminished pancreatic insulin secretion, dyslipidemia and increased atherosclerotic coronary heart disease risk Citation[4,11]. During negative caloric balance, lipolysis may exceed lipogenesis. Circulating free fatty acids may therefore be increased, particularly during the initial period of active and substantial weight loss Citation[8]. This may help explain why free fatty acids may increase with short-term dexfenfluramine use (which is also associated with increased free fatty acid turnover and oxidation) Citation[84]. Conversely, other short-term studies suggest dexfenfluramine decreases circulating free fatty acids Citation[85], which may be related to increased insulin sensitivity associated with weight reduction. The free fatty acid effect of long-term 5-HT agonism is not well described. While a reduction in circulating free fatty acids is a potentially important (chronic) therapeutic effect of anti-obesity interventions , the effect of lorcaserin on free fatty acids has yet to be reported.
Pathogenic adipose tissue endocrine responses
Adipose tissue is an active endocrine organ Citation[9,86–88]. Adiposopathy results in a number of pathogenic endocrine responses that contribute to metabolic disease Citation[3,5,6]. Interventions that improve pathogenic adipose tissue endocrine responses improve metabolic disease Citation[12,13]. Dexfenfluramine administration may result in favorable endocrine effects, such as a decrease in insulin levels Citation[85]. The effect of lorcaserin on some of the more commonly recognized pathogenic adipose tissue endocrine factors has yet to be reported .
Pathogenic adipose tissue immune responses
Adipose tissue is an active immune organ Citation[89,90]. Adiposopathy results in a number of pathogenic immune responses that contribute to metabolic disease Citation[3,5,6]. Interventions that also improve pathogenic adipose tissue immune responses improve metabolic disease Citation[12,13]. Animal studies suggest that fenfluramine may decrease anti-inflammatory markers such as TNF and proinflammatory interleukins; however the mechanism as to why this might occur is unclear Citation[91]. The effect of lorcaserin on some of the more commonly recognized pathogenic adipose tissue immune factors has yet to be reported .
Interaction & ‘cross talk’ with other body organs
Pathogenic endocrine and immune adipose tissue responses do not act alone in causing metabolic disease. Instead, the degree to which pathogenic adipose tissue responses result in metabolic disease is highly dependent upon the interaction and ‘cross talk’ with other body organs such as the nervous system, immune system, skeletal muscle, cardiovascular system, liver, gastrointestinal system, adrenal cortex and thyroid Citation[11]. It is likely that the main beneficial effect of lorcaserin on metabolic disease is mediated through a reduction in appetite, decrease in caloric intake, and the subsequent reduction in adiposity and adiposopathy. Lorcaserin has other theoretical second-order signaling effects that could conceivably affect other body organs . However, any contribution of lorcaserin second-order signaling in improving metabolic disease, independent of the effects upon adipose tissue, has yet to be reported.
Safety & tolerability
Historical perspective of 5-HT receptor agonist safety
As previously noted, fenfluramine underwent clinical evaluation in the 1960s, and received US approval for use as an anti-obesity agent in 1973. The dextrostereoisomer of fenfluramine, dexfenfluramine, received approval in 1996. While serotonin levels were not increased by these agents, cardiac serotonergic activity was increased through stimulation of heart and heart valve 5-HT2b receptors, especially by the norfenfluramine metabolite Citation[92]. While both were known to be associated with rare cases of primary pulmonary hypertension, it was the unexpected finding of cardiac valvulopathy that, in 1997, resulted in the ‘voluntary withdrawal’ of both fenfluramine and dexfluramine – a withdrawal that was in response to a request made by the US FDA.
The association between nonselective 5-HT receptor agonists and heart valve abnormalities was first published in the New England Journal of Medicine in 1997, through a listing of case reports of 24 women without known history of cardiac disease who received the combination of fenfluramine and phentermine Citation[93]. In the ‘Background’ section of the publication, the authors noted the potential scope of the safety concern:
“Although the combination has not been approved by the FDA, in 1996, the total number of prescriptions in the United States for fenfluramine and phentermine exceeded 18 million.”
The authors then acknowledged the ‘serendipitous’ nature of this finding in the ‘Methods’ section, when describing how the individual case reports were found and reported:
“All the patients were identified during the course of routine evaluation for various clinical problems. No attempt was made to identify patients by reviewing databases, conducting cross-index searches of patient files, or soliciting reports of suspected cases from clinical practices. As increasing numbers of patients were identified with similar clinical features, a perceived association between these features and previous or current use of fenfluramine–phentermine evolved. The serendipitous connection between these individual cases was identified as a result of communication among several physicians beginning in May 1996.”
In other words, the data prompting the withdrawal of fenfluramine and dexfenfluramine was not derived from prospective, controlled, clinical trial data, but rather from a listing of case reports and subsequent retrospective reviews. Furthermore, while these authors did report that the cardiac valvular lesions were identical to egotamine-induced and carcinoid valve disease, and while fenfluramine was known to act through serotonergic pathways, the authors acceded to a lack of certainty regarding the potential mechanism in stating: “the precise process by which this might occur is not known”. Even as late as 2005, when describing fenfluramine and dexfenfluramine, one author stated Citation[26]:
“Within a year of its approval, dexfenfluramine was being dispensed at a rate of 85,000 prescriptions per week. Within a year and a half of its approval, the drug was off the market, as was fenfluramine. Reports implicated the 2 drugs in a wave unusual cases of left-sided valvular degeneration – a risk that no one saw coming, and to this day, one that eludes a biomechanistic explanation.”
Despite the unclear mechanism as to why these nonselective 5-HT receptor agonists caused heart valve disease, subsequent retrospective analyses of patients administered fenfluramine and dexfluramine also supported serotonin-associated valvulopathy Citation[94–97], which was characterized by thickening and fibrosis of heart valves. Owing to the different approaches in identifying cases, the reported prevalence of cardiac valvular regurgitation with these agents was widely variable, ranging between 6 and 33% Citation[98]. One small, noncontrolled analysis examined patients who had previous echocardiograms performed from 1993 to 1995 while participating in a weight-loss program, and who received treatment with fenfluramine or dexfenfluramine in combination with mazindol or phentermine. Once fenfluramine and dexfluramine were withdrawn in 1997, these weight-loss program participants were invited to return for a follow-up echocardiogram. This study found that, among those who agreed to return, and who had no valvular regurgitation on previous echocardiograms, 16% (13 out of 79) had ‘new’ valvular regurgitation upon obtaining the follow-up echocardiogram. The valvulopathy appeared to be dependent upon duration, with no new cases of valvular regurgitation in patients exposed to fenfluramine or dexfenfluramine for less than 8 months Citation[99].
Irrespective of the implications from these retrospective and uncontrolled studies, definitive conclusions were difficult to reach without the availability of hard data derived from large, prospective, blinded, randomized, controlled, confirmatory clinical trials. Nonetheless, evidence at the time suggested that the most specific heart valve abnormality associated with nonselective 5-HT receptor agonists was mild-to-more severe aortic regurgitation. The prevalence of aortic regurgitation of dexfenfluramine monotherapy, or fenfluramine in combination with phentermine appeared to be approximately 0–4%, respectively, when administered for 3 months or less, 8–13% when administered for 3 months or longer, and 14–16% when administered for over 1 year Citation[100,13].
The accuracy of such figures derived from nonprospective data was made even more difficult because aortic regurgitation can be found in obese patients, even without use of anti-obesity drugs Citation[101]. It was, therefore, thought reasonable to be diligent in the meticulous evaluation of cardiac valvular abnormalities in the development of any new 5-HT receptor agonist, given that:
• Cardiac valvular abnormalities were associated with both fenfluramine and dexfenfluramine Citation[100];
• Cardiac valvular abnormalities were not associated with phentermine monotherapy (which was approved in 1959 Citation[26], and is the most widely prescribed anti-obesity agent);
• Cardiac valvular abnormalities associated with nonselective serotonin (5-HT) receptor agonists were identical to those found with carcinoid syndrome (defined as a collection of symptoms due to increased serotonin levels produced by gastrointestinal and lung tumors);
• Similar cardiac valvular abnormalities were known to be induced by other pharmaceuticals whose mechanisms include serotoninergic pathways Citation[102].
Paradoxically, the revelation of previously unknown, unsuspected, yet critical safety findings may have presented strategic advantages in the development of novel 5-HT receptor agonists. Investigators now knew where to focus regarding safety surveillance. Fenfluramine was in widespread clinical use in the 1960s through to the 1990s. Dexfenfluramine was highly prescribed in 1996. Their withdrawal was not due to a lack of efficacy or lack of tolerability. Instead, they were withdrawn due to rare, specific, yet critical safety findings that could reasonably be assessed in future, prospective, clinical trials of related investigational agents.
Lorcaserin safety & tolerability: Phase II clinical trial data
In the 12-week lorcaserin clinical trial described previously, lorcaserin was generally well tolerated with the most frequent adverse experiences (AEs) being transient headache, nausea and ‘dizziness’. Among the more unique aspects of this Phase II study was the degree of focus upon heart valve and pulmonary artery pressure (PAP) safety evaluations Citation[49]. In addition to cardiovascular history, physical exam and electrocardiogram assessments performed at study visits, this clinical trial also included echocardiograms performed at screening and at the end of the study (day 85), or study exit. A pool of three qualified cardiologists at a central echocardiography core laboratory read the echocardiograms, and all were blinded to study subject identifiers. Each sonographer was given a detailed protocol, and required to interpret all images according to standard criteria. The same cardiologists, who were blinded to the treatment groups, read all echocardiograms for a given subject and included side-by-side evaluation of initial and end-of-study echocardiograms. Intravenous agitated saline was infused to optimize the evaluation of systolic PAP. Through the interpretation of almost 800 echocardiograms conducted within the framework of a protocol specifically designed to minimize reader bias, the conclusion was that lorcaserin showed no demonstrable evidence of drug-related effects on heart valves or PAP. These safety findings were consistent with the theory that the underlying cause of cardiac valvulopathy by fenfluramine and dexfenfluramine was the nonselective stimulation of the cardiac and heart valve 5-HT2b receptor Citation[92,103–106]. It was also consistent with lorcaserin being a highly selective 5-HT2c receptor agonist with 100-fold functional selectivity versus 5-HT2B receptors Citation[49,48].
Lorcaserin efficacy: Phase III clinical trial data
In 2006, a much larger and more extensive Phase III lorcaserin development program began. The three sentinel Phase III trials included: Behavioral Modification and Lorcaserin for Overweight and Obesity Management Trial (BLOOM); Behavioral Modification and Lorcaserin Second Study for Obesity Management (BLOSSOM); and Behavioral Modification and Lorcaserin for Overweight and Obesity Management in Diabetes Mellitus (BLOOM-DM). Through more thorough assessments of glucose metabolism, blood pressure, lipids and other parameters associated with metabolic disease, these trials will help better define the degree to which lorcaserin not only improves the weight of patients, but also improves the metabolic health of patients. Collectively, these trials will involve thousands of patients undergoing clinical cardiovascular assessments, supported by tens of thousands of echocardiograms. As such, these clinical trials will provide much longer term clinical trial data regarding lorcaserin’s tolerability and safety.
Conclusion
Lorcaserin 10 mg twice a day promotes significant weight loss in obese patients. Lorcaserin also improves adiposopathy-related metabolic parameters such as glucose metabolism, blood pressure and lipid levels. Thus far, lorcaserin clinical trials have demonstrated no objective evidence of cardiac valvulopathy. Lorcaserin, therefore, represents an anti-obesity agent that not only improves adiposity, but also likely improves adiposopathy with potential improvements in the metabolic health of patients.
Expert commentary & five-year view
The following is a speculation as to how lorcaserin, as a representative of a safe and effective anti-obesity agent, might best fit into a future adiposity and adiposopathy treatment paradigm.
Firstly, it is possible that the term ‘obesity’ will soon become obsolete, except to the extent that it might describe excessive fat-mass-related morbidities. The degree of adiposity that contributes to the onset and/or worsening of metabolic disease varies substantially among individuals and patient populations Citation[3,11]. A substantial range of weight reduction is required to improve metabolic disease in the individual overweight patient. This suggests that far better diagnostic criteria are needed to determine the relationship between adiposity and adiposopathy than simply height and weight (BMI) Citation[81]. But before diagnostic criteria for the pathogenic potential of adipose tissue (adiposopathy) can be established, clinical researchers, ‘opinion leaders’ and scientific organizations must better accept that adipose tissue has pathogenic potential, at least to the extent that is known by basic scientists, clinicians and especially by patients.
Basic science data derived from countless preclinical animal and human studies support the pathogenic effects of adipose tissue, and its contribution to metabolic disease Citation[3,5,9]. In the day-to-day practice of medicine, clinicians well-know that increases in bodyweight increases the risk of metabolic disease in their patients. This clinical experience is supported by objective data Citation[11], and is the impetus as to why many clinicians often encourage overweight patients with metabolic disease to lose weight. In fact, the official regulatory indication for most metabolic drug therapies is as an ‘adjunct’ to appropriate nutritional therapy and increased physical activity. Similarly, many patients are cognizant that weight gain is unhealthy. Many also understand that if they are overweight and have metabolic disease, they will likely become healthier if they lose weight. Unfortunately, many challenges to the pathogenic potential of adipose tissue exist Citation[6], and no scientific organization has yet established diagnostic criteria as to when adiposity might reasonably account for adiposopathy.
This is unsustainable, given the epidemic nature of both adiposity and associated metabolic disease. To illustrate this point, it is acknowledged that a Type 2 diabetes mellitus patient who weighs 300 lbs (with an ideal bodyweight of 150 lbs) may develop diabetes mellitus eye disease, such as disease of the retina (a thin layer of cells lining much of the orbit). The adipose tissue in this patient likely weighs over 150 lbs, and represents this patient’s largest organ by weight. It seems unreasonable to readily accept the pathogenic potential of a tissue measured in microns (retinopathy), yet deny the pathogenic potential of an organ that can be measured in hundreds of pounds, and that may constitute over half of the patient’s bodyweight. Clinicians would therefore benefit from well-founded diagnostic criteria Citation[2] that better allow them to determine when an increase in bodyweight most increases the risk of glucose, blood pressure and lipid abnormalities, and when a reduction in bodyweight might reasonably be expected to improve them. Once such diagnostic criteria are established, then therapeutic interventions used to treat excessive body fat might better be characterized by their ability to improve the (metabolic) health of patients, rather than the degree to which they reduce the weight of patients. Furthermore, once adiposopathy diagnostic criteria are established, then treatment indications might then be established that would better allow greater access to therapies that improve metabolic disease, even if the patient is only modestly overweight. Finally, establishing diagnostic criteria and treatment indications for adiposopathy will likely prompt greater investigation, development, and subsequent regulatory approval of agents that not only reduce adiposity, but also improve multiple metabolic parameters in dysmetabolic overweight patients, even if the improvements in individual metabolic parameters with the agent would not otherwise be sufficient to currently obtain an approvable treatment indication Citation[107].
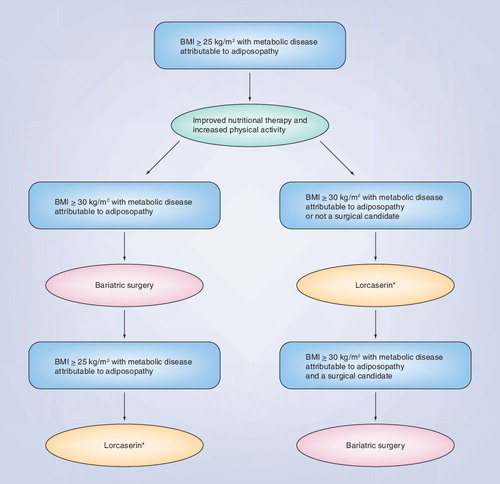
Another example of ‘recognizing the obvious’ Citation[10] as an approach towards improving the health of overweight patients is to acknowledge that public health and ‘diet’ initiatives, however well-intentioned and however effective in an individual patient, have failed to reverse the ‘obesity epidemic’ Citation[202]. Furthermore, given all the redundant and recalcitrant body systems that prioritize avoidance of starvation, it is unlikely that a single pill or even a combination of pills will cause the 300-lb patient previously described to achieve an ideal bodyweight. In the not too distant future, bariatric surgery may become recognized as the treatment of choice for significantly overweight patients, which is an approach that some might argue is already most consistent with an objective assessment of current clinical trial data Citation[8]. If advancement in science continues to support these concepts, if these concepts become accepted, and if lorcaserin is approved, then the following may be better supported:
• Adipose tissue has no less pathogenic potential than the ‘opathies’ of other body organs Citation[13];
• Adiposopathy is a better treatment target to improve metabolic disease in overweight patients than adiposity alone Citation[81];
• Just as with the ‘opathies’ of numerous other body organs, diagnostic criteria for adiposopathy need to be established Citation[2];
• Just as with the ‘opathies’ of numerous other body organs, treatment indications for adiposopathy need to be established Citation[107];
• Bariatric surgery not only improves the weight of patients, but also improves the metabolic health of patients, with effectiveness in patients with a BMI greater than 30 kg/m2Citation[8];
• Bariatric surgery techniques will continue to improve with regard to safety, such that undergoing bariatric surgery will become viewed as being no more unusual (and performed no less frequently) than other common surgeries, such as gall bladder or hernia surgery Citation[8,108];
• Improved surgical techniques, improved surgical efficacy, improved surgical safety, and continued data regarding reduction in overall mortality will make bariatric surgery increasingly more cost effective Citation[8];
• Adiposity and adiposopathy drug therapy, such as lorcaserin, will best be indicated for markedly overweight patient with metabolic disease who are not surgical candidates, or for modestly overweight patients with adiposopathy wherein a reduction in 5–10% of bodyweight would be expected to improve metabolic parameters associated with metabolic disease. This may prove to include patients with metabolic disease who have BMI as low as 25–30 kg/m2.
Given these ‘speculations,’ describes a conceivable future algorithm for the treatment of adiposopathy.
Table 1. Adipogenic and adipose tissue metabolic effects of illustrative CNS factors promoted by 5-HT2c agonism.
Table 2. Examples of treatments for adiposopathy and their effects upon illustrative and selected adipose tissue factors that may contribute to metabolic disease.
Box 1. Adiposopathy (‘sick fat’): summary of causality, as well as examples of anatomical, pathophysiological and clinical manifestations.
Causes of adiposopathy
• Positive caloric balance Citation[5]
• Sedentary lifestyle Citation[5]
• Genetic predisposition, such as in Asian Indians and Pima Indians Citation[3]
• Environmental causes, such as hypercortisolemia Citation[3,5,12]
Anatomical manifestations of adiposopathy
• Adipocyte hypertrophy Citation[3]
• Visceral adiposity Citation[3]
• Growth of adipose tissue beyond its vascular supply
• Ectopic fat deposition in other body organs Citation[4]
Pathophysiologic manifestations of adiposopathy
• Impaired adipogenesis, such as impaired adipocyte proliferation and/or differentiation Citation[3]
• Increased circulating free fatty acids Citation[4]
• Pathogenic adipose tissue endocrine responses, for example increased leptin, increased TNFα, decreased adiponectin, and increased mineralocorticoids Citation[3,6,8]
• Pathogenic adipose tissue immune responses, for example a net increase in proinflammatory responses through increased TNFα and decreased adiponectin Citation[3].
Pathogenic interactions or pathogenic ‘cross talk’ with other body organs, for example liver, muscle and CNSCitation[11]
Clinical manifestations of adiposopathyCitation[3,6]
• Hyperglycemia
• High blood pressure
• Dyslipidemia
• Metabolic syndrome
• Atherosclerosis
• Fatty liver
• Hyperandrogenemia in women
• Hypoandrogenemia in men
• Cancer
Key issues
• Lorcaserin is a selective 5-hydroxytryptamine (serotonin) 2c (5-HT2c) receptor agonist that improves adiposity by promoting weight loss.
• Through reduction in adiposity and waist circumference, lorcaserin improves adiposopathy, and thus improves multiple metabolic parameters associated with metabolic disease.
• Lorcaserin is generally well-tolerated, with the most common adverse experiences being transient headache, nausea and ‘dizziness’.
• Thus far, clinical trial results support that lorcaserin has no adverse effects on heart valves or pulmonary artery pressures.
• Through second-order signaling, lorcaserin may have theoretical benefits on adipose tissue metabolism, and thus theoretical benefits in treating metabolic disease that may be independent of weight loss. However, available evidence suggests that the main improvement in adipose tissue function is through its promotion of weight loss.
• Lorcaserin’s effectiveness in treating patients with diabetes mellitus, hypertension and dyslipidemia (as well as epilepsy, obsessive–compulsive disorders, Parkinson’s disease, schizophrenia, depression, anxiety, sleep disorders and drug abuse) await clinical trials, or the results of clinical trials in these specific patient populations.
• Based upon available evidence, lorcaserin appears to be a generally safe and well-tolerated agent that not only improves the weight of patients, but also improves the metabolic health of patients.
Financial & competing interests disclosure
Harold Bays has served as a consultant, and received research grants from Arena Pharmaceuticals. He has also served as an advisor for regulatory matters concerning drug development, been a primary investigator for over 400 clinical trials, and served as a consultant and speaker for multiple pharmaceutical companies. The author has no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript
Notes
Adiposity and obesity can also result in diseases related to fat mass, as well as diseases related to fat dysfunction (adiposopathy).
References
- Kushner RF, Roth JL. Assessment of the obese patient. Endocrinol. Metab. Clin. North Am.32(4), 915–933 (2003).
- Bays H, Abate N, Chandalia M. Adiposopathy: sick fat causes high blood sugar, high blood pressure, and dyslipidemia. Future Cardiol.1(1), 39–59 (2005).
- Bays HE, Gonzalez-Campoy JM et al. Pathogenic potential of adipose tissue and metabolic consequences of adipocyte hypertrophy and increased visceral adiposity. Expert Rev. Cardiovasc. Ther.6(3), 343–368 (2008).
- Bays H, Mandarino L, DeFronzo RA. Role of the adipocyte, free fatty acids, and ectopic fat in pathogenesis of Type 2 diabetes mellitus: peroxisomal proliferator-activated receptor agonists provide a rational therapeutic approach. J. Clin. Endocrinol. Metab.89(2), 463–478 (2004).
- Bays H, Ballantyne C. Adiposopathy: why do adiposity and obesity cause metabolic disease? Future Lipidol.1(4), 389–420 (2006).
- Bays HE, Gonzalez-Campoy JM, Henry RR et al. Is adiposopathy (sick fat) an endocrine disease? Int. J. Clin. Pract.62(10), 1474–1483 (2008).
- Khan T, Muise ES, Iyengar P et al. Metabolic dysregulation and adipose tissue fibrosis: role of collagen VI. Mol. Cell. Biol.29(6), 1575–1591 (2009).
- Bays HE, Laferrere B, Dixon J et al. Adiposopathy and bariatric surgery: is ‘sick fat’ a surgical disease? A consensus from the adiposopathy and bariatric surgery group. Int. J. Clin. Pract.(2009) (In Press).
- Bluher M. Adipose tissue dysfunction in obesity. Exp. Clin. Endocrinol. Diabetes117(6), 241–250 (2009).
- Bays H. Adiposopathy, metabolic syndrome, quantum physics, general relativity, chaos and the Theory of Everything. Expert Rev. Cardiovasc. Ther.3(3), 393–404 (2005).
- Bays HE. ‘Sick fat,’ metabolic disease, and atherosclerosis. Am. J. Med.122(1 Suppl.), S26–S37 (2009).
- Bays H, Blonde L, Rosenson R. Adiposopathy: how do diet, exercise, weight loss and drug therapies improve metabolic disease? Expert Rev. Cardiovasc. Ther.4(6), 871–895 (2006).
- Bays HE, Rodbard RW, Schorr AB, González-Campoy JM. Adiposopathy: treating pathogenic adipose tissue to reduce cardiovascular disease risk. Curr. Treat Options Cardiovasc. Med.9(4), 259–271 (2007).
- Bays HE, Chapman RH, Grandy S. The relationship of body mass index to diabetes mellitus, hypertension and dyslipidaemia: comparison of data from two national surveys. Int. J. Clin. Pract.61(5), 737–747 (2007).
- Lavie CJ, Milani RV, Ventura HO. Obesity and cardiovascular disease: risk factor, paradox, and impact of weight loss. J. Am. Coll. Cardiol.53(21), 1925–1932 (2009).
- Davenport DL, Xenos ES, Hosokawa P, Radford J, Henderson WG, Endean ED. The influence of body mass index obesity status on vascular surgery 30-day morbidity and mortality. J. Vasc. Surg.49(1), 140–147, 147 (2009).
- Orpana HM, Berthelot JM, Kaplan MS, Feeny DH, McFarland B, Ross NA. BMI and mortality: results from a national longitudinal study of canadian adults. Obesity (Silver.Spring)DOI: 10.1038/oby.2009.191 (2009) (Epub ahead of print).
- Karelis AD, St-Pierre DH, Conus F, Rabasa-Lhoret R, Poehlman ET. Metabolic and body composition factors in subgroups of obesity: what do we know? J. Clin. Endocrinol. Metab.89(6), 2569–2575 (2004).
- Tsai AG, Wadden TA. Systematic review: an evaluation of major commercial weight loss programs in the United States. Ann. Intern. Med.142(1), 56–66 (2005).
- Rubino F. Is Type 2 diabetes an operable intestinal disease? A provocative yet reasonable hypothesis. Diabetes Care31(Suppl. 2), S290–S296 (2008).
- Patel MR, Donahue M, Wilson PW, Califf RM. Clinical trial issues in weight-loss therapy. Am. Heart J.151(3), 633–642 (2006).
- Bays H. Adiposopathy: the endocannabinoid system as a therapeutic treatment target for dysfunctional ‘sick’ fat. CJHP19(1), 32–39 (2007).
- Hausenloy DJ. Contrave TM: novel treatment for obesity. Clin. Lipidol.4(3), 279–285 (2009).
- Gadde KM, Yonish GM, Foust MS, Wagner HR. Combination therapy of zonisamide and bupropion for weight reduction in obese women: a preliminary, randomized, open-label study. J. Clin. Psychiatry68(8), 1226–1229 (2007).
- Gadde KM, Yonish GM, Foust MS, Tam PY, Najarian T. A 24-week randomized controlled trial of VI-0521, a combination weight loss therapy, in obese adults. Presented at: Program and abstracts of the Annual Scientific Scientific Meeting of NAASO, The Obesity Society, 2006. Boston, MA, USA, 20–24 October 2006.
- Colman E. Anorectics on trial: a half century of federal regulation of prescription appetite suppressants. Ann. Intern. Med.143(5), 380–385 (2005)
- Greenway FL, Caruso MK. Safety of obesity drugs. Expert Opin. Drug Saf.4(6), 1083–1095 (2005).
- Snow V, Barry P, Fitterman N, Qaseem A, Weiss K. Pharmacologic and surgical management of obesity in primary care: a clinical practice guideline from the American College of Physicians. Ann. Intern. Med.142(7), 525–531 (2005).
- Varady KA, Tussing L, Bhutani S, Braunschweig CL. Degree of weight loss required to improve adipokine concentrations and decrease fat cell size in severely obese women. Metabolism58(8), 1096–1101 (2009).
- Ratner RE. An update on the Diabetes Prevention Program. Endocr. Pract.12(Suppl. 1), 20–24 (2006).
- Nair RP, Ren J. Pharmacotherapy of obesity–benefit, bias and hyperbole. Curr. Med. Chem.16(15), 1888–1897 (2009).
- Bays H, Dujovne C. Anti-obesity drug development. Expert Opin. Investig. Drugs11(9), 1189–1204 (2002).
- Bays HE. Current and investigational antiobesity agents and obesity therapeutic treatment targets. Obes. Res.12(8), 1197–1211 (2004).
- Magni P, Dozio E, Ruscica M et al. Feeding behavior in mammals including humans. Ann. NY Acad. Sci.1163, 221–232 (2009).
- Lenard NR, Berthoud HR. Central and peripheral regulation of food intake and physical activity: pathways and genes. Obesity (Silver.Spring)16(Suppl. 3) S11–S22 (2008).
- Garfield AS, Heisler LK. Pharmacological targeting of the serotonergic system for the treatment of obesity. J. Physiol.587(Pt 1), 49–60 (2009).
- Akana SF. Feeding and stress interact through the serotonin 2C receptor in developing mice. Physiol. Behav.94(4), 569–579 (2008).
- Scalfi L, D’Arrigo E, Carandente V, Coltorti A, Contaldo F. The acute effect of dexfenfluramine on resting metabolic rate and postprandial thermogenesis in obese subjects: a double-blind placebo-controlled study. Int. J. Obes. Relat. Metab. Disord.17(2), 91–96 (1993).
- Van Gaal LF, Vansant GA, Steijaert MC, De Leeuw I. Effects of dexfenfluramine on resting metabolic rate and thermogenesis in premenopausal obese women during therapeutic weight reduction. Metabolism44(2 Suppl. 2), 42–45 (1995).
- Lafreniere F, Lambert J, Rasio E, Serri O. Effects of dexfenfluramine treatment on body weight and postprandial thermogenesis in obese subjects. A double-blind placebo-controlled study. Int. J. Obes. Relat. Metab. Disord.17(1), 25–30 (1993).
- Kogon MM, Krauchi K, van der Velde V, Van der Werf H, Keller U. Psychological and metabolic effects of dietary carbohydrates and dexfenfluramine during a low-energy diet in obese women. Am. J. Clin. Nutr.60(4), 488–493 (1994).
- Doucet E, St Pierre S, Almeras N, Mauriege P, Richard D, Tremblay A. Changes in energy expenditure and substrate oxidation resulting from weight loss in obese men and women: is there an important contribution of leptin? J. Clin. Endocrinol. Metab.85(4), 1550–1556 (2000).
- Levitsky DA, Troiano R. Metabolic consequences of fenfluramine for the control of body weight. Am. J. Clin. Nutr.55(1 Suppl), S167– S172 (1992).
- Halford JC, Harrold JA, Boyland EJ, Lawton CL, Blundell JE. Serotonergic drugs: effects on appetite expression and use for the treatment of obesity. Drugs67(1), 27–55 (2007).
- Recasens MA, Barenys M, Sola R, Blanch S, Masana L, Salas-Salvado J. Effect of dexfenfluramine on energy expenditure in obese patients on a very-low-calorie-diet. Int. J. Obes. Relat. Metab. Disord.19(3), 162–168 (1995).
- Wacker DA, Miller KJ. Agonists of the serotonin 5-HT2C receptor: preclinical and clinical progression in multiple diseases. Curr. Opin. Drug Discov. Devel.11(4), 438–445 (2008).
- Miller KJ. Serotonin 5-ht2c receptor agonists: potential for the treatment of obesity. Mol. Interv.5(5), 282–291 (2005).
- Thomsen WJ, Grottick AJ, Menzaghi F et al. Lorcaserin, a novel selective human 5-hydroxytryptamine2C agonist: in vitro and in vivo pharmacological characterization. J. Pharmacol. Exp. Ther.325(2), 577–587 (2008).
- Smith SR, Prosser W, Donahue D, Anderson C, Shanahan W. APD356–004 study group. Lorcaserin (APD356), a selective 5-HT2C agonist, safely induces weight loss in a 12-week study of healthy obese patients. Presented at: American Diabetes Association 66th Annual Scientific Sessions. Washington DC USA, 6 December 2006.
- Sugden K, Tichopad A, Khan N, Craig IW, D’Souza UM. Genes within the serotonergic system are differentially expressed in human brain. BMC Neurosci.10, 50 (2009).
- Kaumann AJ, Levy FO. 5-hydroxytryptamine receptors in the human cardiovascular system. Pharmacol. Ther.111(3), 674–706 (2006).
- Xu J, Jian B, Chu R, Lu Z et al. Serotonin mechanisms in heart valve disease II: the 5-HT2 receptor and its signaling pathway in aortic valve interstitial cells. Am. J. Pathol.161(6), 2209–2218 (2002).
- Ross EM, Roberts WC. The carcinoid syndrome: comparison of 21 necropsy subjects with carcinoid heart disease to 15 necropsy subjects without carcinoid heart disease. Am. J. Med.79(3), 339–354 (1985).
- Moss N, Choi Y, Cogan D et al. A new class of 5-HT2B antagonists possesses favorable potency, selectivity, and rat pharmacokinetic properties. Bioorg. Med. Chem. Lett.19(8), 2206–2210 (2009).
- Shyu KG. Serotonin 5-HT2B receptor in cardiac fibroblast contributes to cardiac hypertrophy: a new therapeutic target for heart failure? Circ. Res.104(1), 1–3 (2009).
- Xu Y, Jones JE, Kohno D et al. 5-HT2CRs expressed by pro-opiomelanocortin neurons regulate energy homeostasis. Neuron60(4), 582–589 (2008).
- Lam DD, Przydzial MJ, Ridley SH, Yeo GS, Rochford JJ, O’Rahilly S, Heisler LK. Serotonin 5-HT2C receptor agonist promotes hypophagia via downstream activation of melanocortin 4 receptors. Endocrinology149(3), 1323–1328 (2008).
- Morgan M, Chen W, Anderson C, Prosser W, Donahue D, Shanahan W. Pharmacokinetic properties, metabolism, and tolerability of lorcaserin in health volunteers. Presented at: Annual Meeting of The Obesity Society. Phoenix, AZ, USA, 3–7 October 2008.
- Munro JF, Seaton DA, Duncan LJ. Treatment of refractory obesity with fenfluramine. Br. Med. J.2(5514), 624–625 (1966).
- Stahl KA, Imperiale TF. An overview of the efficacy and safety of fenfluramine and mazindol in the treatment of obesity. Arch. Fam. Med.2(10), 1033–1038 (1993).
- Salmela PI, Sotaniemi EA, Viikari J, Solakivi-Jaakkola T, Jarvensivu P. Fenfluramine therapy in non-insulin-dependent diabetic patients: effects on body weight, glucose homeostasis, serum lipoproteins, and antipyrine metabolism. Diabetes Care4(5), 535–540 (1981).
- Hudson KD. The anorectic and hypotensive effect of fenfluramine in obesity. JR. Coll. Gen. Pract.27(181), 497–501 (1977).
- O’Connor HT, Richman RM, Steinbeck KS, Caterson ID. Dexfenfluramine treatment of obesity: a double blind trial with post trial follow up. Int. J. Obes. Relat. Metab. Disord.19(3), 181–189 (1995).
- Turner P. Dexfenfluramine. Its place in weight control. Drugs39(Suppl. 3), 53–62 (1990).
- Carvajal A, García del Pozo J, Martín de Diego I, Rueda de Castro AM, Velasco A. Efficacy of fenfluramine and dexfenfluramine in the treatment of obesity: a meta-analysis. Methods Find. Exp. Clin. Pharmacol.22(5), 285–290 (2000).
- Weintraub M, Hasday JD, Mushlin AI, Lockwood DH. A double-blind clinical trial in weight control. Use of fenfluramine and phentermine alone and in combination. Arch. Intern. Med.144(6), 1143–1148 (1984).
- Weintraub M, Sundaresan PR, Schuster B et al. Long-term weight control study. II (weeks 34 to 104). An open-label study of continuous fenfluramine plus phentermine versus targeted intermittent medication as adjuncts to behavior modification, caloric restriction, and exercise. Clin. Pharmacol. Ther.51(5), 595–601 (1992).
- Weintraub M, Sundaresan PR, Schuster B, Moscucci M, Stein EC. Long-term weight control study. III (weeks 104 to 156). An open-label study of dose adjustment of fenfluramine and phentermine. Clin. Pharmacol. Ther.51(5), 602–607 (1992).
- Weintraub M, Sundaresan PR, Schuster B et al. Long-term weight control study. IV (weeks 156 to 190). The second double-blind phase. Clin. Pharmacol. Ther.51(5), 608–614 (1992).
- Weintraub M, Sundaresan PR, Schuster B, Averbuch M, Stein EC, Byrne L. Long-term weight control study. V (weeks 190 to 210). Follow-up of participants after cessation of medication. Clin. Pharmacol. Ther.51(5), 615–618 (1992).
- Weintraub M, Sundaresan PR, Schuster B. Long-term weight control study. VII (weeks 0 to 210). Serum lipid changes. Clin. Pharmacol. Ther.51(5), 634–641 (1992).
- Weintraub M. Long-term weight control study: conclusions. Clin. Pharmacol. Ther.51(5), 642–646 (1992).
- Smith BM, Smith JM, Tsai JH et al. Discovery and structure-activity relationship of (1R)-8-chloro-2,3,4,5-tetrahydro-1-methyl-1H-3-benzazepine (Lorcaserin), a selective serotonin 5-HT2C receptor agonist for the treatment of obesity. J. Med. Chem.51(2), 305–313 (2008).
- Grundy SM, Cleeman JI, Daniels SR et al. Diagnosis and management of the metabolic syndrome: an American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement. Circulation112(17), 2735–2752 (2005).
- Dattilo AM, Kris-Etherton PM. Effects of weight reduction on blood lipids and lipoproteins: a meta-analysis. Am. J. Clin. Nutr.56(2), 320–328 (1992).
- Aubin D, Gagnon A, Grunder L, Dent R, Allen M, Sorisky A. Adipogenic and antiapoptotic protein levels in human adipose stromal cells after weight loss. Obes. Res.12(8), 1231–1234 (2004).
- Liese AD, Mayer-Davis EJ, Tyroler HA et al. Development of the multiple metabolic syndrome in the ARIC cohort: joint contribution of insulin, BMI, and WHR. Atherosclerosis risk in communities. Ann. Epidemiol.7(6), 407–416 (1997).
- Janssen I, Katzmarzyk PT, Ross R. Waist circumference and not body mass index explains obesity-related health risk. Am. J. Clin. Nutr.79(3), 379–384 (2004).
- Zhu S, Heshka S, Wang Z, Shen W, Allison DB, Ross R, Heymsfield SB. Combination of BMI and waist circumference for identifying cardiovascular risk factors in whites. Obes. Res.12(4), 633–645 (2004).
- Blackburn GL, Waltman BA. Pharmacotherapy to reduce visceral fat. Clin. Cornerstone7(2–3), 52–60 (2005).
- Bays H, Dujovne CA. Adiposopathy is a more rational treatment target for metabolic disease than obesity alone. Curr. Atheroscler. Rep.8(2), 144–156 (2006).
- Grundy SM. Obesity, metabolic syndrome, and cardiovascular disease. J. Clin. Endocrinol. Metab.89(6), 2595–2600 (2004).
- Swinburn BA, Carmichael HE, Wilson MR. Dexfenfluramine as an adjunct to a reduced-fat, ad libitum diet: effects on body composition, nutrient intake and cardiovascular risk factors. Int. J. Obes. Relat. Metab. Disord.20(11), 1033–1040 (1996).
- Greco AV, Mingrone G, Capristo E, De, Gaetano A., Ghirlanda G, Castagneto M. Effects of dexfenfluramine on free fatty acid turnover and oxidation in obese patients with Type 2 diabetes mellitus. Metabolism44(2 Suppl. 2), 57–61 (1995).
- Ditschuneit HH, Flechtner-Mors M, Dolderer M, Fulda U, Ditschuneit H. Endocrine and metabolic effects of dexfenfluramine in patients with android obesity. Horm. Metab. Res.25(11), 573–578 (1993).
- Miner JL. The adipocyte as an endocrine cell. J. Anim. Sci.82(3), 935–941 (2004).
- Kershaw EE, Flier JS. Adipose tissue as an endocrine organ. J. Clin. Endocrinol. Metab.89(6), 2548–2556 (2004).
- Ahima RS. Adipose tissue as an endocrine organ. Obesity.(Silver Spring)14(Suppl. 5) S242–S249 (2006).
- Caspar-Bauguil S, Cousin B, Galinier A et al. Adipose tissues as an ancestral immune organ: site-specific change in obesity. FEBS Lett.579(17), 3487–3492 (2005).
- Schaffler A, Muller-Ladner U, Scholmerich J, Buchler C. Role of adipose tissue as an inflammatory organ in human diseases. Endocr. Rev.27(5), 449–467 (2006).
- Connor TJ, Kelly JP. Fenfluramine-induced immunosuppression: an in vivo analysis. Eur. J. Pharmacol.455(2–3), 175–185 (2002).
- Rothman RB, Baumann MH. Serotonergic drugs and valvular heart disease. Expert Opin. Drug Saf.8(3), 317–329 (2009).
- Connolly HM, Crary JL, McGoon MD et al. Valvular heart disease associated with fenfluramine-phentermine. N. Engl. J. Med.337(9), 581–588 (1997).
- Centers for Disease Control and Prevention (CDC). Cardiac valvulopathy associated with exposure to fenfluramine or dexfenfluramine: US Department of Health and Human Services interim public health recommendations, November 1997. MMWR Morb. Mortal Wkly Rep.46, 1061–1066 (1997).
- Connolly HM, Crary JL, McGoon MD et al. Valvular heart disease associated with fenfluramine-phentermine. N. Engl. J. Med.337(24), 1783 (2009).
- Sachdev M, Miller WC, Ryan T, Jollis JG. Effect of fenfluramine-derivative diet pills on cardiac valves: a meta-analysis of observational studies. Am. Heart J.144(6), 1065–1073 (2002).
- Weissman NJ, Tighe JF Jr, Gottdiener JS, Gwynne JT. An assessment of heart-valve abnormalities in obese patients taking dexfenfluramine, sustained-release dexfenfluramine, or placebo. Sustained-Release Dexfenfluramine Study Group. N. Engl. J. Med.339(11), 725–732 (1998).
- Jollis JG, Landolfo CK, Kisslo J, Constantine GD, Davis KD, Ryan T. Fenfluramine and phentermine and cardiovascular findings: effect of treatment duration on prevalence of valve abnormalities. Circulation101(17), 2071–2077 (2000).
- Ryan DH, Bray GA, Helmcke F et al. Serial echocardiographic and clinical evaluation of valvular regurgitation before, during, and after treatment with fenfluramine or dexfenfluramine and mazindol or phentermine. Obes. Res.7(4), 313–322 (1999).
- Gardin JM, Schumacher D, Constantine G, Davis KD, Leung C, Reid CL. Valvular abnormalities and cardiovascular status following exposure to dexfenfluramine or phentermine/fenfluramine. JAMA283(13), 1703–1709 (2000).
- Gardin JM, Constantine G, Davis K, Leung C, Reid CL. Aortic valvular regurgitation: prevalence and clinical characteristics in a predominantly obese adult population not taking anorexigens. Echocardiography23(7), 569–576 (2006).
- Smith SA, Waggoner AD, Fuentes LD, vila-Roman VG. Role of serotoninergic pathways in drug-induced valvular heart disease and diagnostic features by echocardiography. J. Am. Soc. Echocardiogr.22(8), 883–889 (2009).
- Fitzgerald LW, Burn TC, Brown BS et al. Possible role of valvular serotonin 5-HT(2B) receptors in the cardiopathy associated with fenfluramine. Mol. Pharmacol.57(1), 75–81 (2000).
- Rothman RB, Baumann MH, Savage JE et al. Evidence for possible involvement of 5-HT(2B) receptors in the cardiac valvulopathy associated with fenfluramine and other serotonergic medications. Circulation102(23), 2836–2841 (2000).
- Mekontso-Dessap A, Brouri F et al. Deficiency of the 5-hydroxytryptamine transporter gene leads to cardiac fibrosis and valvulopathy in mice. Circulation113(1), 81–89 (2005).
- Roth BL. Drugs and valvular heart disease. N. Engl. J. Med.356(1), 6–9 (2007).
- Bays H. Adiposopathy–defining, diagnosing, and establishing indications to treat ‘sick fat’: what are the regulatory considerations? US Endocrine Disease2, 12–14 (2006).
- Flum DR, Belle SH, King WC et al. Perioperative safety in the longitudinal assessment of bariatric surgery. N. Engl. J. Med.361(5), 445–454 (2009).
- Krude H, Biebermann H, Schnabel D et al. Obesity due to proopiomelanocortin deficiency: three new cases and treatment trials with thyroid hormone and ACTH4–10. J. Clin. Endocrinol. Metab.88(10), 4633–4640 (2003).
- Obregon MJ. Thyroid hormone and adipocyte differentiation. Thyroid18(2), 185–195 (2008).
- Levacher C, Sztalryd C, Kinebanyan MF, Picon L. Effects of thyroid hormones on adipose tissue development in Sherman and Zucker rats. Am. J. Physiol.246(1 Pt 1), C50–C56 (1984).
- Bays H. The melanocortin system as a therapeutic treatment target for adiposity and adiposopathy. Drugs RD7(5), 289–302 (2006).
- Nogueiras R, Wiedmer P, Perez-Tilve D et al. The central melanocortin system directly controls peripheral lipid metabolism. J. Clin. Invest.117(11), 3475–3488 (2007).
- Raposinho PD, White RB, Aubert ML The melanocortin agonist Melanotan-II reduces the orexigenic and adipogenic effects of neuropeptide Y (NPY) but does not affect the NPY-driven suppressive effects on the gonadotropic and somatotropic axes in the male rat. J. Neuroendocrinol.15(2), 173–181 (2003).
- Kernie SG, Liebl DJ, Parada LF. BDNF regulates eating behavior and locomotor activity in mice. EMBO J.19(6), 1290–1300 (2000).
- Bradley RL, Mansfield JP, Maratos-Flier E, Cheatham B. Melanin-concentrating hormone activates signaling pathways in 3T3-L1 adipocytes. Am. J. Physiol. Endocrinol. Metab.283(3), E584–E592 (2002).
- Digby JE, Chen J, Tang JY, Lehnert H, Matthews RN, Randeva HS. Orexin receptor expression in human adipose tissue: effects of orexin-A and orexin-B. J. Endocrinol.191(1), 129–136 (2006).
Websites
- Centers for Disease Control and Prevention. Overweight and Obesity http://cdc.gov/obesity/index.html (Accessed 20 July 2009).
- World Health Organization. Global Strategy on Diet, Physical Activity and Health www.who.int/dietphysicalactivity/publications/facts/obesity/en/ (Accessed 19 November 2009)
- Precription Medications for the Treatment of Obesity. US Department of Health and Human Resources.National Institutes of Health.http://win.niddk.nih.gov/publications/PDFs/prescriptionmeds1104bw.pdf (Accessed 9 September 2009)
- Yu B, Nichols CD. Serotonin 5-HT2A receptor activity mediates adipocyte differentiation. The Journal of the Federation of American Societies for Experimental Biology. www.fasebj.org/cgi/content/meeting_abstract/23/1_MeetingAbstracts/941.5 (Accessed 8 August 2009).