Abstract
This article reviews important presentations from the 7th Screening Europe Conference, and extracts general trends and developments. Technological advances, as well as novel applications are discussed, thus providing an overview on state-of-the-art high-throughput screening. Among other topics, cell-based assays were highly popular, since they reflect the clinical situation much more closely than screens based on purified drug targets. A further approach to reduce the costly attrition of drug candidates subsequent to initial screens is the use of label-free technology. By measuring desired properties directly, without the use of any reporter (e.g., calorimetric measurements of binding affinities), the selection of false positives can be decreased drastically. Additional improvements in high-throughput screening are resulting from novel technology platforms utilizing sophisticated imaging systems and/or miniaturization. These and other important topics from the Screening Europe 2010 Conference are discussed in this article, thus providing a current snapshot of the field.
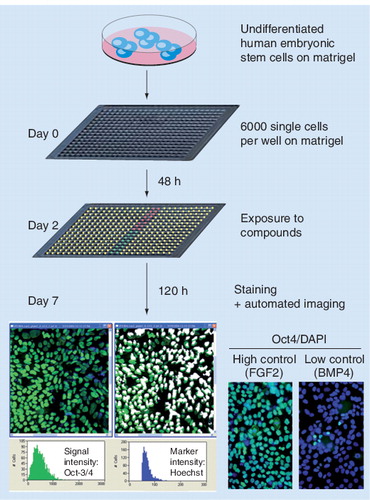
The cells were exposed to a total of 2800 different chemical entities and subsequently stained for Oct4 expression as a marker for the undifferentiated state. Reproduced courtesy of Sabrina Desbordes, Center for Genomic Regulation, Barcelona, Spain.
HEK293T cells expressing a fluorescence reporter for cell viability are directly cocultivated with the pathogen of interest (Staphylococcus aureus). (A) In the presence of a noncytotoxic, specific antibiotic (e.g., penicillin/streptomycin), the human reporter cells survive and generate a strong readout signal (inset); whereas, (B) in the absence of a specific antibiotic, the pathogen outgrows the human reporter cells, resulting in shutdown of the readout signal Citation[10].
![Figure 2. Screening compounds simultaneously for the inhibition of bacterial growth and cytotoxicity in human cells.HEK293T cells expressing a fluorescence reporter for cell viability are directly cocultivated with the pathogen of interest (Staphylococcus aureus). (A) In the presence of a noncytotoxic, specific antibiotic (e.g., penicillin/streptomycin), the human reporter cells survive and generate a strong readout signal (inset); whereas, (B) in the absence of a specific antibiotic, the pathogen outgrows the human reporter cells, resulting in shutdown of the readout signal Citation[10].](/cms/asset/1ae4b907-f3fe-474a-99a6-5fca9bc6abe1/iero_a_11214286_f0002_b.jpg)
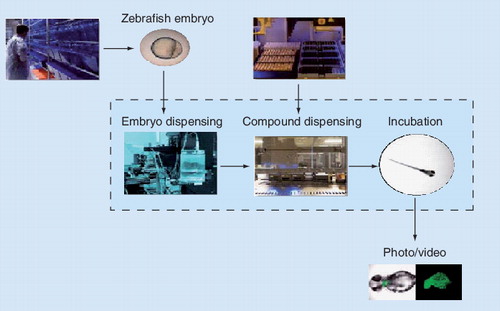
Zebrafish embryos show 85% genetic homology with humans and develop rapidly. This allows the screening of several thousand samples at costs similar to cell-based screens.
Reproduced courtesy of Carlos Callol-Massot, Biobide S.L., San Sebastian, Spain.
The Screening Europe Conference is the biggest of its kind on European ground. It is held annually, with participants from both academic institutions and industry. Hence, it is a good platform for exchange between scientists focusing on basic research and applied science. This year, approximately 70 speakers were presenting their latest findings, complemented by another 69 posters and an exhibition of companies focusing on screening applications. This article reviews general trends in the field, as illustrated in a number of the many excellent presentations of the conference.
Cell-based assays
In general, the vast majority of compounds, identified as ‘hits’ during biochemical screening, fail in the later stages of development, leading to very costly attrition of selected compounds Citation[1]. It is desirable, therefore, to find out more about the biology of a hit compound as early as possible and one potential way to do this is using cell-based assays. Indeed, it is highly unlikely that any lead will progress to become a drug candidate without having first demonstrated activity in a cell-based model. When cell-based assays are used to select hits, many properties of the tested compound are detected, in addition to the desired inhibitory effect on pathogenesis. Cytotoxicity, cell permeability (in case the target biomolecule is intracellular) and effects on cell growth can be monitored at the same time. Furthermore, libraries of compounds can be tested without having an identified target, sometimes leading to the identification of new drug targets involved in the pathology. In a keynote presentation, Richard Eglen (Bio-discovery, Perkin Elmer, MA, USA) pointed out that drug screens to preclinical animal testing can be performed without having identified the drug target Citation[2], as long as the desired phenotype is clearly defined. Furthermore, he presented novel screening approaches based on primary cells (e.g., peripheral blood mononuclear cells [PBMCs]), taking cell heterogeneity into account and, thus, being even closer to the clinical situation Citation[3,4]. However, the biological variability caused approximately 10–20% failure of the assay.
Darren Cawkill (Pfizer, Sandwich, UK) presented different approaches for the generation of cell lines (stably or transiently) expressing the desired drug target Citation[5]. Prior to a screen for novel drugs, the expression level and the response to characterized drugs must be analyzed. This can be done in an automated fashion using plate- or fluorescence-activated cell-sorting (FACS)-based assays. In a case study, Cawkill presented the screening of cell clones expressing transient receptor potential (TRP) channels using the Cello™ system (The Automation Partnership, Royston, UK). During the primary screen, 266 out of 960 clones were selected for high gene expression. Ultimately, only three clones passed the two subsequent screens (electrophysiology and binding assay), and were analyzed in detail: 99.7% of the population expressed measurable levels of TRP channels, compared with just 35% for a previously generated cell line. This not only demonstrates the variability of biological systems, but also the need for automated monitoring and selection of cell clones prior to a given drug screen.
In initial studies, stem cells were applied to high-throughput small-molecule screens . Sabrina C Desbordes (Center for Genomic Regulation, Barcelona, Spain) presented a screen for compounds controlling self-renewal and differentiation of human embryonic stem cells Citation[6,7]. The assay was based on Oct4 expression as a marker for nondifferentiated cells, and proved to be reliable (Z-factor of 0.37). Using libraries of marketed drugs and natural compounds (Prestwick and MicroSource libraries), a total of 2800 different chemical entities were screened. In consequence, compounds promoting short-term human embryonic stem cell (hESC) maintenance (e.g., theanine and flurbiprofen) were identified, as well as compounds directing early lineage choice during differentiation (e.g., tretinoin and sarmentogenin).
In general, high-throughput screens do not have to be based on a single-cell species. The demand for a better understanding of the drug mechanism and possible side effects has made screens using multiple-cell types increasingly popular. This can be done by either testing the effect of compounds on different cell lines in parallel, or by using, cocultures. Igor Ivanov (Onkolead, Munich, Germany) presented an approach based on the screening of drugs on a panel of 80 different cell types, for which whole-genome-expression profiles are available Citation[8]. This way, the effect of drugs on different targets can be monitored comprehensively. In an alternative approach, Martin Augustin (Millipore Corporation, Munich, Germany) presented screens for efficacy and safety profiling based on coculturing different cell types in the same sample Citation[9]. Neurons and astrocytes were incubated together in the presence of different drug candidates, and applied to high-content imaging. The subsequent staining of nuclei, βIII-tubulin, synaptophysin and glial fibrillary acidic protein, allowed neurotoxicity profiles for each compound to be recorded. Obviously, this kind of high-content screen reveals significantly more data than conventional cytotoxicity assays based on a single readout (e.g., methyl tetrazolium or lactate dehydrogenase assays) for a single-cell line and, thus, allows comprehensive efficacy and safety profiling.
Monitoring cytotoxic effects during the primary screen can also be achieved in indirect inhibition assays. In this case, pathogens, such as viruses, bacteria or even cancer cells, are directly cocultivated with human reporter cells . These cells generate a strong fluorescence signal solely in the presence of compounds that specifically inhibit the pathogen without affecting their own viability Citation[10]. Hence, compounds can be screened simultaneously for therapeutic and cytotoxic effects Citation[11,12].
Although assays based on individual cell types or cocultures facilitate the monitoring of adverse side effects, the ultimate test for cytotoxicity is the administration of a given drug candidate to a living animal. Depending on the model species, this step can be performed in a high-throughput fashion. Carles Callot-Massot (Biobide S.L., San Sebastian, Spain) presented toxicity screens in zebrafish embryos (Danio rerio), having 85% genetic homology with humans Citation[13]. Since the embryos are transparent and show rapid development and organogenesis, they are ideally suited for high-throughput high-content microscopic analyses . Up to several thousand embryos can be monitored in a single screen, at costs similar to those of cell-based assays. Furthermore, ethical issues are decreased drastically in comparison with conventional animal testing. Nonetheless, even the impact of compounds on the heart (e.g., causing bradycardia, arrhythmia or even ventricle failure) can be assessed easily.
Label-free & fragment-based screening
In addition to the selection of cytotoxic compounds, assay artefacts can lead to the costly attrition of drug candidates during later stages of development or (pre)clinical trials. One way to reduce this phenomenon is the application of label-free technology. Instead of analyzing a reporter signal, such as cellular fluorescence (which, for example, can be biased by the autofluorescence of dead cells), a physical property of the drug candidate (e.g., binding) is measured directly using surface plasmon resonance (SPR), calorimetry, nuclear magnetic resonance (NMR) or crystallography. In a keynote presentation, Frederik Sundberg (GE Healthcare, Uppsala, Sweden) pointed out the conceptual advantages of SPR technology: native biomolecules and compounds can be screened at very low target consumption Citation[14]. Jörg Bomke (Merck Serono, Darmstadt, Germany) added that commercial systems have reached sensitivity and throughput for the screening of medium-sized libraries, and presented a case study in which 1920 fragments were assayed Citation[15]. Jeffrey Albert (Astra Zeneca, DE, USA) presented a similar approach (based on SPR, NMR and crystallography) and defined fragments as compounds with a molecular weight of 120–300 Da, less than three hydrogen bond donors/acceptors, and a partition coefficient in octanol/water (cLogP) of less than 3 Citation[16]. The advantage of fragment-based screening is the fact that vastly fewer screening candidates are required to explore chemical structure space Citation[17]. Although hits in the primary screen usually show binding affinities and IC50 values in the low millimolar range, subsequent structural optimization enables the development of potent drugs. In a case study, Albert presented the discovery of a β-secretase inhibitor with an IC50 value of 80 nM. A possible drawback of fragment-based screening is the requirement for high compound concentrations (due to the low initial affinities), which might result in the selection of unspecific binders during the primary screen, for which reason, counter screens are absolutely essential.
Screening for selectivity
Drugs that bind or inhibit their target in a rather unspecific way must be ruled out as early as possible during the screening process, since they might cause undesired side effects. To do so, the selectivity of a hit must be analyzed quantitatively. Joost Uitdehaag (Merck Research Laboratories, Oss, The Netherlands) introduced the ‘selectivity entropy’ as a scoring system for specificity Citation[18]. When incubating inhibitor molecules with different drug targets, they distribute according to their binding preference and, hence, selection entropies can be determined. Highly specific inhibitors will show a very narrow distribution (bind to only one of multiple potential drug targets) and, thus, show very low entropy values. In comparison with the previous scoring systems for selectivity, such as the Gini coefficient Citation[19] or the Karaman selectivity score Citation[20], the selectivity entropy is a nonarbitrary measure that is independent of substrate and inhibitor concentrations. To illustrate the value of this concept, Uitdehaag pointed out that low-selectivity entropies correlate well with low attrition rates during clinical trials.
Selectivity is also of major importance for receptor ligands. Many receptor (ant)agonists not only mediate the desired therapeutic effect, but also cause some severe side effects. Strikingly, the beneficial and adverse effects are not necessarily mediated by the same downstream signal pathways (of the same receptor). So-called biased ligands have been identified, selectively interfering with particular pathways Citation[21,22]. Focusing on free fatty acid receptors, Graeme Milligan (University of Glasgow, Scotland, UK) demonstrated the importance of using different assays in parallel Citation[23]. In this way, the differential activation of downstream effectors can be analyzed in detail. Furthermore, time-resolved measurements can be extremely useful, if the different pathways show different kinetics.
Novel screening & assay platforms
Recent progress in synthesis technology and genomics has drastically increased the demand for novel screening platforms. To improve throughput and decrease the assay costs, minimal assay volumes are highly desirable. Therefore, droplet-based microfluidic systems have been presented, in which aqueous droplets (pico- to nano-liter volumes) of a water-in-oil emulsion serve as independent reaction vessels Citation[10]. The tiny assay volumes not only give rise to massive cost savings (>1000-fold), but also facilitate screens on the single-cell level, or the screening of samples that can hardly be generated on the scale for conventional high-throughput screening (e.g., primary cells). The technology enables the quantitative analysis and sorting of up to 1.8 × 106 samples/h Citation[24,25], and even allows the recording of kinetic data Citation[26].
Richard Ellson (Labcyte, CA, USA) presented microtiter plate systems, in which liquids are transferred by acoustic droplet ejection Citation[27]. A transducer below the well of a microtiter plate generates a strong sound wave, expelling a droplet from the meniscus of the sample. Subsequently, the droplet travels through the air into the well of a receiving microtiter plate placed above, in upside-down orientation (the samples in this plate are held by capillary forces). This approach enables the transfer of very small sample volumes (5–50 nl with a coefficient of variation [CV] of <3%) without the need for pipette tips or pins (possibly causing carry-over between different samples of a plate). Potential problems are caused by varying contact angles at the meniscus (water–air interface), owing to proteins or surfactants in the samples. However, this limitation can be overcome by adjusting the power of the sound wave dynamically.
A quite similar system was discussed by Tobias Brode and colleagues (Fraunhofer Institute, Stuttgart, Germany). Their ‘immediate drop on-demand technology (i-doT)’ is based on the transfer of droplets by a pressure pulse Citation[28]. A microtiter plate with a little orifice (110 µm in diameter) in each well is placed above the target samples (receiving microtiter plate). Subsequently, a millisecond pressure pulse (greater than the capillary pressure sealing the orifice) is applied, resulting in the ejection of a single droplet (4–7 nl with a CV of ∼1.5%), rapidly entering the target well. This technology is even compatible with cell suspensions and allows the transfer of more than 100,000 samples per day.
Larry Sklar (University of New Mexico, NM, USA) presented a system for multiplex target screening by high-throughput flow cytometry Citation[29]. A so-called HyperCyt® device aspirates samples (volume of 2 µl) from microtiter plates and loads them into a length of tubing, where they are stably separated by air bubbles. Subsequently, the sample arrays are injected into a FACS, where they are analyzed at a rate of 40 samples/min. This approach will be instrumental in the NIH Roadmap Molecular Libraries Initiative.
Novel drug targets
In a keynote presentation, Peter McNaughton (University of Cambridge, UK) introduced two recently characterized TRP ion channels, and their modulators, as novel drug targets Citation[30]. They trigger the increased heat-pain sensation upon injury or inflammation, an effect termed inflammatory hyperalgesia. McNaughton showed that TRPV1 has a proalgesic effect, and is sensitized by inflammatory mediators, while TRPM8 has an analgesic effect and is inhibited by inflammatory mediators. These findings should be of major interest for a new class of painkillers.
Wendy Lea (NIH Chemical Genomics Center, MD, USA) presented a high-throughput screening approach for the discovery of novel anesthetics with reduced side effects Citation[31]. Her system makes use of a new surrogate marker for the binding to the neurotransmitter GABA, a ligand-gated ion channel that can hardly be overexpressed and, thus, shows very poor abundancy. However, recent studies demonstrated that the binding of drug candidates to the well-expressed horse spleen apoferritin, closely mirrors the inhibition of GABA Citation[32]. Lea and colleagues exploited this observation and identified 13 new compounds with IC50 values in the micromolar range.
Conclusion
The 7th Screening Europe Conference was an excellent opportunity for getting a snapshot of the latest high-throughput screening technologies and applications. Avoiding the costly attrition of hits during later stages of drug development has become a major topic in the field. Hence, screening campaigns have become more and more complex, not only to monitor efficacy, but also cytotoxicity and selectivity. This demand for high-throughput high-content data drives the development of novel screening technology and assays, a development from which the patient could, ultimately, profit, in the form of novel drugs with minimized side effects.
Financial & competing interests disclosure
The author is an inventor on patents related to assays and technologies described in this article and could potentially benefit from corresponding revenues according to the inventor reward schemes of several academic institutions. The author has no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Related Research Data
References
- Bleicher KH, Böhm HJ, Müller K, Alanine AI. Hit and lead generation: beyond high-throughput screening. Nat. Rev. Drug Discov.2(5), 369–378 (2003).
- Feng Y, Mitchison TJ, Bender A, Young DW, Tallarico JA. Multi-parameter phenotypic profiling: using cellular effects to characterize small-molecule compounds. Nat. Rev. Drug Discov.8(7), 567–578 (2009).
- Eglen RM. Primary, iPS and stem cells: emerging tools for HTS? Presented at: 7th Annual Screening Europe Conference. Barcelona, Spain, 11–12 February 2010.
- Eglen RM, Gilchrist A, Reisine T. An overview of drug screening using primary and embryonic stem cells. Comb. Chem. High Throughput Screen.11(7), 566–572 (2008).
- Cawkill D. Cell based assay development – it all starts with the cell. Presented at: 7th Annual Screening Europe Conference. Barcelona, Spain, 11–12 February 2010.
- Desbordes SC. High-throughput screening in human embryonic stem cells for drug discovery. Presented at: 7th Annual Screening Europe Conference. Barcelona, Spain, 11–12 February 2010.
- Desbordes SC, Placantonakis DG, Ciro A et al. High-throughput screening assay for the identification of compounds regulating self-renewal and differentiation in human embryonic stem cells. Cell Stem Cell2(6), 602–12 (2008).
- Ivanov I. sciWIFTER applications for in vitro cellular screening. Presented at: 7th Annual Screening Europe Conference. Barcelona, Spain, 11–12 February 2010.
- Augustin M. Technology spotlight: illuminating your pathway to discovery: cell based assays for efficacy and safety profiling. Presented at: 7th Annual Screening Europe Conference. Barcelona, Spain, 11–12 February 2010.
- Merten CA. Indirect inhibition assays and droplet-based microfluidics. Presented at: 7th Annual Screening Europe Conference. Barcelona, Spain, 11–12 February 2010.
- Granieri L, Miller OJ, Griffiths AD, Merten CA. A competition-based assay for the screening of species-specific antibiotics. J. Antimicrob. Chemother.64(1), 62–68 (2009).
- Clausell-Tormos J, Griffiths AD, Merten CA. Coupling the inhibition of viral transduction with a positive fluorescence signal. Comb. Chem. High Throughput Screen.13(4), 352–357 (2010).
- Callol-Massot C. Validation of a zebrafish automated screening platform. Presented at: 7th Annual Screening Europe Conference. Barcelona, Spain, 11–12 February 2010.
- Sundberg F. Using label-free technologies to improve screening and hit validation. Presented at: 7th Annual Screening Europe Conference. Barcelona, Spain, 11–12 February 2010.
- Bomke J. Rapid, label-free screening and affinity determination of fragments binding specifically to drug targets. Presented at: 7th Annual Screening Europe Conference. Barcelona, Spain, 11–12 February 2010.
- Albert J. Challenges and successes in fragment-based lead generation: increasing impact across drug discovery. Presented at: 7th Annual Screening Europe Conference. Barcelona, Spain, 11–12 February 2010.
- Teague SJ, Davis AM, Leeson PD, Oprea T. The design of leadlike combinatorial libraries. Angew. Chem. Int. Ed. Engl.38(24), 3743–3748 (1999).
- Uitdehaag J. Screening for selectivity. Presented at: 7th Annual Screening Europe Conference. Barcelona, Spain, 11–12 February 2010.
- Graczyk PP. Gini coefficient: a new way to express selectivity of kinase inhibitors against a family of kinases. J. Med. Chem.50(23), 5773–5779 (2007).
- Karaman MW, Herrgard S, Treiber DK et al. A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol.26(1), 127–132 (2008).
- Walters RW, Shukla AK, Kovacs JJ et al. β-arrestin1 mediates nicotinic acid-induced flushing, but not its antilipolytic effect, in mice. J. Clin. Invest.119(5), 1312–1321 (2009).
- Taggart AK, Kero J, Gan X et al. (D)-β-hydroxybutyrate inhibits adipocyte lipolysis via the nicotinic acid receptor PUMA-G. J. Biol. Chem.280(29), 26649–26652 (2005).
- Milligan SE. What are the right assays to select? Studies with receptors for free fatty acids. Presented at: 7th Annual Screening Europe Conference. Barcelona, Spain, 11–12 February 2010.
- Clausell-Tormos J, Lieber D, Baret JC et al. Droplet-based microfluidic platforms for the encapsulation and screening of mammalian cells and multicellular organisms. Chem. Biol.15(5), 427–437 (2008).
- Sklar L. Multiplex target screening by high throughput flow cytometry. Presented at: 7th Annual Screening Europe Conference. Barcelona, Spain, 11–12 February 2010.
- Granieri L, Baret J-C, Griffiths AD, Merten CA. High-throughput screening of enzymes by retroviral display using droplet-based microfluidics. Chem. Biol.17(3), 229–235 (2010).
- Clausell-Tormos J, Griffiths AD, Merten CA. An automated two-phase microfluidic system for kinetic analyses and the screening of compound libraries. Lab. Chip DOI: 10.1039/b921754a (2010) (Epub ahead of print).
- Ellson R. Total assay assembly enabled with acoustic droplet ejection. Presented at: 7th Annual Screening Europe Conference. Barcelona, Spain, 11–12 February 2010.
- Brode T. i-doT (immediate drop on demand technology) fast and direct sample handling for MWPs. Presented at: 7th Annual Screening Europe Conference. Barcelona, Spain, 11–12 February 2010.
- McNaughton P. Thermo-TRP ion channels as drug targets. Presented at: 7th Annual Screening Europe Conference. Barcelona, Spain, 11–12 February 2010.
- Lea W. A high-throughput approach for identification of candidate general anesthetics. Presented at: 7th Annual Screening Europe Conference. Barcelona, Spain, 11–12 February 2010.
- Vedula LS, Brannigan G, Economou NJ et al. A unitary anesthetic binding site at high resolution. J. Biol. Chem.284(36), 24176–24184 (2009).
Website
- NIH Roadmap Molecular Libraries Initiative http://nihroadmap.nih.gov