Abstract
Patients with von Hippel–Lindau disease (VHL) often harbor significant disease burden within the CNS, specifically craniospinal-axis hemangioblastomas and endolymphatic sac tumors (ELSTs). The majority (60–80%) of patients with VHL harbor hemangioblastomas, and 10–15% will develop ELSTs. Advances in the understanding of the natural history and outcomes associated with the surgical management of VHL-associated tumors have led to improved management of patients with VHL. Optimizing indications for surgical intervention and refining of surgical techniques for these lesions can reduce patient morbidity associated with the management of this syndrome. In this article, we review the various aspects of perioperative management of patients with VHL, surgical indications and general operative principles for the management of hemangioblastomas and ELSTs, and outcomes associated with the surgical treatment of these tumors.
Medscape: Continuing Medical Education Online
This activity has been planned and implemented in accordance with the Essential Areas and policies of the Accreditation Council for Continuing Medical Education through the joint sponsorship of Medscape, LLC and Expert Reviews Ltd. Medscape, LLC is accredited by the ACCME to provide continuing medical education for physicians.
Medscape, LLC designates this Journal-based CME activity for a maximum of 1 AMA PRA Category 1 Credit(s)™. Physicians should claim only the credit commensurate with the extent of their participation in the activity.
All other clinicians completing this activity will be issued a certificate of participation. To participate in this journal CME activity: (1) review the learning objectives and author disclosures; (2) study the education content; (3) take the post-test with a 70% minimum passing score and complete the evaluation at http://www.medscape.org/journal/expertneurothera; (4) view/print certificate.
Release date: September 28, 2011; Expiration date: September 28, 2012
Learning objectives
Upon completion of this activity, participants should be able to:
• Describe the perioperative management of patients with CNS lesions of VHL
• Describe the management of hemangioblastomas in patients with VHL
• Describe the management of ELST in patients with VHL
Financial & competing interests disclosure
EDITOR
Elisa Manzotti,Editorial Director, Future Science Group, London, UK
Disclosure:Elisa Manzotti has disclosed no relevant financial relationships.
CME AUTHOR
Laurie Barclay, MD,Freelance writer and reviewer, Medscape, LLC
Disclosure:Laurie Barclay, MD, has disclosed no relevant financial relationships.
AUTHORS
Joshua J Wind, MD,Surgical Neurology Branch, National Institute of Neurological Disorders and Stroke, NIH, Bethesda, MD, USA; and Department of Neurological Surgery, George Washington University Medical Center, Washington, DC, USA
Disclosure:Joshua J Wind has disclosed no relevant financial relationships.
Russell R Lonser, MD,Surgical Neurology Branch, National Institute of Neurological Disorders and Stroke, NIH, Bethesda, MD, USA
Disclosure:Russell R Lonser has disclosed no relevant financial relationships.
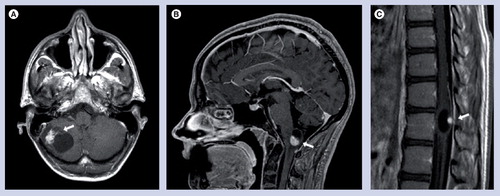
Postcontrast T1-weighted MRIs of (A) a cerebellar, (B) brainstem and (C) spinal cord hemangioblastoma. Arrows in each image indicate the location of the hemangioblastoma mural nodule, with its associated peritumoral cyst.
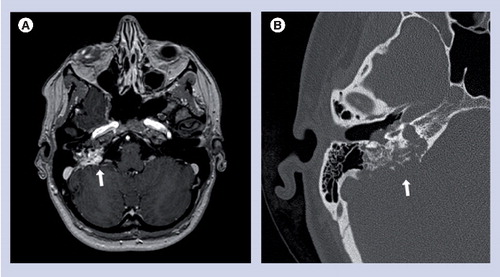
(A) Axial postcontrast T1-weighted MRI and (B) axial noncontrast high-resolution CT scan through the right temporal bone demonstrating endolymphatic sac tumor (arrows) with bony erosion of the petrous temporal bone.
von Hippel–Lindau disease (VHL) is a multiple organ system neoplastic syndrome and is transmitted across generations in an autosomal dominant manner. The incidence of VHL is approximately 1 in every 36,000 live births and this genetic disorder has over 90% penetrance Citation[1,2]. The manifestations of VHL are the result of germline mutations in the VHL tumor-suppressor gene on chromosome 3 Citation[3]. VHL manifestations include the kidney, adrenal, pancreatic and reproductive adnexal organs, and CNS lesions. VHL can be diagnosed by clinical criteria (Box 1) and/or genetic testing. Owing to the multisystem effects of VHL, this tumor syndrome requires multidisciplinary management and serial screening Citation[4,5]. Median life expectancy is 49 years Citation[4,6].
CNS manifestations comprise a large portion of the overall tumor burden in VHL patients and underlie significant morbidity and mortality in these patients Citation[2]. CNS VHL-related neoplasms develop primarily below the tentorium and include the cerebellum, brainstem, spinal cord and retinal hemangioblastomas Citation[7], as well as endolymphatic sac tumors (ELSTs). As most patients with VHL will harbor craniospinal hemangioblastomas and/or ELSTs, optimization of the management of these neoplasms is necessary to minimize morbidity and mortality. In this article, we review the features and management strategies for CNS lesions in patients with VHL.
Molecular biology of VHL
The VHL tumor-suppressor gene encodes for the VHL protein, which complexes with other proteins involved in ubiquitin-dependent proteolysis of hypoxia-inducing factor (HIF). Dysregulation of this VHL-associated function causes increased expression of a variety of factors, including erythropoietin, PDGF, VEGF and TGF. Upregulation of these factors may lead to angiogenesis and tumorigenesis Citation[4,8]. Additional mechanisms of tumorigenesis have been described outside of the HIF pathway, including alterations in microtubule binding and stabilization and extracellular matrix composition, as well as apoptotic and transcription regulation Citation[8].
Perioperative management of VHL patients
The multisystem effects of VHL result in potentially complicating visceral factors associated with the management of CNS lesions in patients with VHL.
Pheochromocytomas
A total of 10–20% of patients with VHL harbor pheochromocytomas. These lesions can present a unique challenge to perioperative management Citation[9]. Patients with VHL are evaluated with 24-h urine collections for catecholamines and plasma-free metanephrines. The combination of these tests can be used to rule out the presence of pheochromocytoma Citation[10,11]. Abdominal and pelvic pre- and post-contrast computed tomography (CT) or MRI are employed to detect adrenal masses, and metaiodobenzylguanidine (MIBG) or PET nuclear imaging can be employed for further evaluation Citation[12]. For patients with VHL who have confirmed or possible pheochromocytomas requiring resection of CNS tumors, preoperative α- and β-adrenergic blockade can be employed to reduce the risk of perioperative hypertensive crisis Citation[13].
Other visceral disease
Renal lesions are present in approximately 60% of patients with VHL, with 24–45% of patients harboring renal cell carcinomas and the remainder renal cysts Citation[2,5]. The urologic management of renal cell carcinoma has evolved to nephron-sparing surgery, with the goals of both decreasing the risk of metastasis while maintaining renal function Citation[14,15]. However, some patients with VHL will present with decreased renal clearance or renal failure, which may complicate imaging and perioperative management. A total of 35–70% of patients with VHL harbor pancreatic neuroendocrine tumors, cysts or cystadenomas Citation[16,17]. Treatment of these lesions may lead to pancreatic insufficiency, including glucose intolerance, which requires judicious management in the perioperative period.
Hemangioblastomas
Tumor features
Hemangioblastomas are benign tumors that may originate from embryologic hemangioblasts Citation[18]. These tumors are highly vascular lesions. The high vascular permeability of these tumors leads to plasma ultrafiltrate leakage into the surrounding interstitium, and once the reabsorptive capacity of the surrounding tissues is surpassed, tissue edema and peritumoral cyst development occurs Citation[19,20]. Symptomatic presentation is often associated with this stage of hemangioblastoma edema and cyst development Citation[21,22].
Hemangioblastomas occur within the cerebellum, brainstem, spinal cord, nerve roots and retina, but rarely above the tentorium. As retinal hemangioblastomas fall outside of the spectrum of this article, we concentrate on reviewing craniospinal hemangioblastoma management. The cerebellum and spinal cord are the most common sites for hemangioblastoma development. In contrast to sporadic hemangioblastomas, which are solitary, patients with VHL frequently have multiple hemangioblastomas along the craniospinal axis Citation[21,22].
Imaging characteristics
Hemangioblastomas enhance avidly on postcontrast CT and T1-weighted MRI sequences. To best delineate peritumoral edema and/or cysts associated with hemangioblastomas, T2-weighted or fluid-attenuated inversion-recovery (FLAIR) magnetic resonance sequences are used. When associated with peritumoral cysts, hemangioblastomas often have the classic ‘cyst with mural nodule’ appearance ; however, these tumors can also less frequently develop complex or intratumoral cysts. If arteriography is employed to image these lesions, it will demonstrate a rich tumor blush with early draining veins and vascular engorgement.
Indications for treatment
Studies delineating the long-term natural history of CNS hemangioblastomas in patients with VHL demonstrate several important features that guide their management Citation[21,22]. Specifically, a saltatory growth pattern is associated with hemangioblastomas, which is characterized by quiescent periods that are interspersed with periods of growth, with quiescent periods lasting an average of over 2 years (mean: 25 months). Despite growth of tumors in nearly all patients, 41% of patients became symptomatic over long-term follow-up (mean follow-up of 12.4 years). Of the patients that did become symptomatic, 45% of symptom-producing tumors were not evident on initial radiographic studies Citation[22].
Owing to the characteristic hemangioblastoma saltatory growth pattern, and until other presymptomatic factors that can predict symptom formation can be identified, these tumors are ideally resected after the development of early signs or symptoms. Based on long-term surveillance data from patients with VHL, if radiographic progression was only used as an indication for hemangioblastoma resection, patients with VHL-associated hemangioblastomas would undergo an average of four additional unneeded operations over a 10-year period. This would expose these patients to operative risk for tumors that would not have become symptomatic Citation[22].
Analysis to determine tumor characteristics that will predict symptom formation has revealed that larger tumors and more rapid growth rates of a tumor and an associated cyst may be associated with symptom development Citation[21,22]. Moreover, previous data demonstrate that peritumoral edema preceeds cyst formation with hemangioblastomas Citation[19,20]. The sequence of edema and cyst development and the association of size and growth rates with symptom development can guide patient education and follow-up in asymptomatic patients.
Operative principles
While arteriography can be helpful in defining vascular anatomy for complex large hemangioblastomas, embolization is not routinely employed as a preoperative adjunct. Hemangioblastomas often derive blood supply from many small vessels in addition to dominant vascular pedicles. Therefore embolization may only be partially effective in decreasing blood flow or intraoperative blood loss. Furthermore, studies have reported significant morbidity (postembolization hemorrhage and infarction) and mortality after embolization Citation[23,24]. The use of newer embolic agents, such as Onyx® (ev3, Covidien Vascular Therapies, Mansfield, MA, USA), has been reported, but warrants further investigation Citation[25].
Regardless of location, the resection of CNS hemangioblastomas is performed using well-defined microsurgical techniques Citation[26–30]. When tumors reach the pial surface, the tumor–pial junction is incised to develop a plane just superficial to the tumor capsule. When the tumor–pial junction cannot be accessed directly or does not reach the pial surface, cortisectomy or myelotomy is required to gain access to the tumor, depending on the location. Following tumor surface identification, circumferential tumor dissection from the surrounding nervous system tissue is performed. Tumor vessels are coagulated and divided individually as they enter the tumor capsule. Upon completion of circumferential dissection, the hemangioblastoma is entirely devascularized and can be removed. In this way, blood loss is avoided by not disrupting the tumor capsule.
When hemangioblastomas have an associated peritumoral cyst, the cyst is maintained intact as long as possible to facilitate tumor dissection. Inspection of the cyst wall is performed after tumor resection to ensure that there are no additional hemangioblastomas contributing to the cyst. With the exception of the case of an intratumoral cyst, the cyst wall of hemangioblastomas is compressed gliotic tissue Citation[19]. The cyst wall is not neoplastic and is not resected, and the cyst will uniformly collapse after the associated hemangioblastoma is resected. Efforts to fenestrate or shunt peritumoral cysts are unnecessary if the underlying tumor is removed.
Resection outcomes
Cerebellar hemangioblastomas
Cerebellar hemangioblastomas account for approximately half of VHL-associated CNS hemangioblastomas; approximately 70% of patients will harbor one or more cerebellar hemangioblastoma Citation[22]. They occur most frequently in the dorsal half of the cerebellar hemispheres, and present with headache, gait difficulties, dysmetria, hydrocephalus and nausea or vomiting. Symptomatic tumors are nearly uniformly associated with either peritumoral edema or peritumoral cysts Citation[26]. Long-term analysis of the outcomes of patients with VHL undergoing resection of cerebellar hemangioblastomas has been performed in 88 patients who underwent 126 operations for 164 cerebellar hemangioblastomas Citation[26]. Immediately after surgery, 88% of patients were improved or stable. At 3 months postoperatively, 98% of patients were improved or stable from their preoperative symptoms. All patients achieved total resection with no tumor recurrence. Hydrocephalus, when present, resolved after tumor resection in 94% of patients without additional intervention, while 6% of patients later required ventriculoperitoneal shunt placement for cerebrospinal fluid diversion Citation[26].
Spinal cord hemangioblastomas
Spinal cord hemangioblastomas account for nearly half of craniospinal hemangioblastomas in VHL Citation[22]. These tumors are evenly distributed between the cervical and thoracic spinal cord. The vast majority of spinal cord hemangioblastomas are dorsal, often involving the dorsal root entry zone (66% of dorsal tumors). Presenting signs and symptoms are most commonly hypesthesia, hyperreflexia, dysesthesia, weakness, proprioceptive deficit and bowel and bladder complaints. Symptomatic tumors are nearly uniformly associated with either peritumoral edema or peritumoral cysts Citation[27,28]. A long-term analysis of the outcomes of 108 patients with VHL undergoing resection of 218 spinal cord hemangioblastomas has been performed. A total of 85% of patients were stable or improved immediately postoperatively. A total of 96% of patients were functionally stable or improved at the 6-month follow-up. The analysis of factors associated with functional deterioration revealed that ventral tumor location or entirely intramedullary location was associated with functional decline postoperatively. Total resection was achieved in nearly all cases (99.5%) Citation[28].
Brainstem hemangioblastomas
Brainstem hemangioblastomas account for 10–15% of craniospinal hemangioblastomas in patients with VHL Citation[31,32]. These tumors are found most frequently at the region of the obex, and present with swallowing difficulties, headache, sensory changes, singultus and impaired gait Citation[29,30]. Symptomatic tumors are most often associated with peritumoral edema and/or cysts Citation[30]. Long-term outcome analysis of 44 patients with VHL who underwent resection of 71 brainstem hemangioblastomas has been performed. A total of 67% of patients maintained their functional status, 16% of patients had improvement and 18% had deterioration postoperatively. Of the patients who deteriorated, 89% returned to their baseline function within 6 months. All patients achieved total resection with no tumor recurrence, and there was no operative mortality Citation[30].
Other locations
Nerve root hemangioblastomas can be found in patients with VHL. Nerve root hemangioblastomas can be intradural extramedullary, extradural or a combination of these. Nerve root hemangioblastomas most often have an intrafascicular nerve root origin and, thus, are frequently not resectable without sacrificing the involved nerve root. Therefore, resection of nerve root hemangioblastomas is reserved for symptomatic tumors with the knowledge that the involved nerve root may have to be sacrificed in order to obtain total tumor resection Citation[33,34].
Hemangioblastomas, while most frequently located within the posterior fossa and spinal cord, can occur anywhere in the craniospinal axis, including the supratentorial compartment in patients with VHL Citation[35,36]. The most frequent supratentorial locations include the pituitary stalk (29% of supratentorial hemangioblastomas), hippocampus (21%) and optic apparatus (14%). As with hemangioblastomas in other locations, surgical intervention is reserved for symptomatic tumors, as these can remain asymptomatic for indefinite periods of time Citation[35].
Radiation therapy
Stereotactic radiosurgery (SRS) has been proposed for the management of hemangioblastomas as a less invasive treatment modality. Short-term retrospective results have documented tumor control rates exceeding 90% Citation[37]. Similarly, prospective long-term assessment of SRS to hemangioblastomas of the cerebellum and brainstem in VHL demonstrate favorable short-term control rates. Alternatively, the long-term prospective results for SRS reveal that control rates drop to 61% at 10 years and 51% at 15 years Citation[38]. Coupled with the saltatory growth pattern of hemangioblastomas, it is possible that short-term success attributed to SRS may be due to periods of tumor quiescence as opposed to actual treatment efficacy. Furthermore, many tumors that are not visible on initial imaging can arise and become symptomatic, illustrating another potential limitation for the use of SRS. SRS may also result in transiently increased peritumoral edema and exacerbate tumor-related symptoms. Fractionated radiotherapy could have a role in the treatment of certain patients with VHL-related hemangioblastomas, but the effectiveness of this radiation modality requires further investigation Citation[39].
Endolymphatic sac tumors
Tumor features
Endolymphatic sac tumors are derived from the endolymphatic epithelium. Specifically, these tumors appear to originate in the vestibular aqueduct endolymphatic epithelium Citation[40]. They are benign tumors that were initially described in patients with VHL in the 1920s Citation[41,42]. However, ELSTs were formally recognized as a component of the VHL syndrome in the late 1990s Citation[43]. In total, 10–15% of patients with VHL harbor ELSTs. As opposed to sporadic ELST cases, ELSTs in patients with VHL can be bilateral in 30% of cases Citation[43–45]. ELSTs present with vestibulocochlear symptoms, including sensorineural hearing loss (95% of patients), tinnitus (90%), episodic vertigo or disequilibrium (66%), and aural fullness (30%). Larger tumors that further invade the petrous bone can cause facial paresis due to facial nerve involvement (8%) Citation[43]. Hearing loss can occur in a sudden, stepwise or more insidious manner Citation[46,47].
There are three potential mechanisms that underlie hearing loss in VHL patients with ELSTs. First, local invasion of the otic capsule by the tumor can lead to disruption of endolymphatic flow and subsequent vestibulopathy and hearing loss. Second, intralabyrinthine hemorrhage may result in a sudden and irreversible hearing loss. Finally, blockage of normal endolymphatic pathways or excessive peritumoral fluid extravasation by the ELST can result in the impairment of normal endolymphatic reabsorption mechansims and hydrops. This can lead to vestibulocochlear dysfunction, including progressive hearing loss Citation[48].
Imaging characteristics
A combination of radiographic modalities is employed to evaluate patients with VHL for ELSTs . High-resolution pre- and post-contrast MRI that includes T1-weighted and FLAIR sequences are routinely used to evaluate for ELSTs. ELSTs may be visualized as an enhancing tumor or asymmetric enhancement of the endolymphatic duct on postcontrast studies (T1-weighted). Tumoral hemorrhage into the labyrinth may be visualized on precontrast T1 and FLAIR sequences. If there is a lesion visualized on MRI or in a patient with concerning vestibulocochlear symptoms, thin-cut temporal bone CT imaging can be used to reveal ELST-associated bony erosion of the vestibular aqueduct. As ELSTs can be microscopic (below the level of MRI detection) at symptom formation, the only imaging finding may be intralabyrinthine hemorrhage Citation[49] or there may be no radiographic findings at all. Subsequently, negative imaging does not rule out an ELST in a patient with VHL.
Audiologic findings
Audiograms in patients with ELSTs often demonstrate decreased speech reception thresholds in affected ears. Hearing loss in these patients can occur abruptly (including in a stepwise, progressive manner), or it can have a more insidious onset. Hearing loss is occasionally responsive to steroid treatment Citation[46].
Indications for surgery
As ELSTs of any size can cause sudden hearing loss due to intralabyrinthine hemorrhage, endolymphatic hydrops or direct otic capsule invasion, we recommend resection of radiographically visible tumors in patients with hearing. In patients without hearing, resection may be recommended when compressive or other neurological (including other audiovestibular) findings are present due to tumor size. We also recommend resection in patients with concerning symptomatology and intralabyrinthine hemorrhage but no visible tumor, as hemorrhage is secondary evidence of a microscopic ELST Citation[46,49].
Operative principles
Endolymphatic sac tumors are hypervascular tumors. ELSTs often take their primary vascular supply from external carotid artery branches Citation[50]. In larger tumors where intraoperative blood loss is a concern, preoperative endovascular embolization can be a helpful adjuvant to decrease tumor vascularity Citation[51]. Very large tumors may also acquire their blood supply from the internal carotid artery.
Multiple surgical approaches have been described and used to resect lesions within the vestibular aqueduct and surrounding region. We routinely employ the retrolabyrinthine posterior petrosectomy (RLPP) to resect small and medium-sized ELSTs. This approach offers excellent anatomic exposure to the sac and duct of the endolymphatic system while allowing for preservation of hearing Citation[46].
Outcomes
In our experience with VHL patients with ELSTs, patients undergo total resection of tumor with preservation of the preoperative level of hearing Citation[46]. Patients may experience a transient 10–20 decibel worsening of hearing in the operated ear postoperatively, which may be due to surgical debris entering the endolymphatic spaces and affecting sound transduction.
Radiation therapy
Given that surgical resection of ELSTs is curative, radiation therapy does not currently have a primary treatment role for tumors amenable to gross total resection. Radiation therapy (stereotactic or fractionated) may be used in the treatment of unresectable ELSTs (those involving the jugular bulb, intrapetrous internal carotid artery or facial nerve) or in patients who are not candidates for surgical resection. Literature on radiation treatment for ELSTs is currently limited to case reports and requires further investigation Citation[52–55].
Expert commentary
Better understanding of the long-range natural history of hemangioblastomas and defined outcomes after surgical management have demonstrated that surgical resection is safe and effective. Surgery is generally reserved for symptomatic lesions given the knowledge of the growth patterns of these tumors and the complexities of tumor management within a neoplasia syndrome such as VHL. Current data does not support the routine prophylactic use of SRS to treat hemangioblastomas in patients with VHL who have surgically accessible tumors and can tolerate surgery.
Surgical resection of ELSTs using the RLPP approach allows the preservation of hearing in VHL patients. Understanding the mechanisms of hearing loss from these tumors and the outcomes associated with their surgical management has led to the recommendation that visible tumors should be resected in patients with hearing in order to prevent future hearing loss. Further studies are required to determine the part that radiation therapy may play in ELST management in patients with VHL.
Five-year view
Further research is currently underway to elucidate radiographic factors that may predict symptom formation in hemangioblastomas. This would allow tumors to be resected prior to symptomatic presentation, while still reserving surgical intervention for only those tumors that would probably become symptomatic. Further research is required to determine the efficacy of fractionated radiation therapy for hemangioblastomas in VHL disease.
Larger scale follow-up is being conducted on a larger series of patients with surgically treated ELSTs, in order to better define outcomes. Radiographic techniques that might demonstrate endolymphatic hydrops in a patient with an otherwise radiographically negative ELST need to be developed and validated. Further research is required to define the role of radiation therapy in ELST management.
Box 1. Clinical criteria for the diagnosis of von Hippel–Lindau disease.
Patients without a family history of von Hippel–Lindau disease
• Two or more CNS hemangioblastomas
or
• One CNS hemangioblastoma and a visceral tumor (excluding epididymal or renal cysts)
Patients with a family history of von Hippel–Lindau disease
• One CNS hemangioblastoma
or
• Pheochromocytoma
or
• Clear cell renal carcinoma
Key issues
• von Hippel–Lindau disease (VHL) is a multisystem neoplastic disorder that requires a multidisciplinary approach for optimum patient management.
• VHL patients have unique perioperative management needs, including pheochromocytomas, diabetes and renal insufficiency.
• Hemangioblastomas occur throughout the craniospinal axis in VHL patients. They exhibit a saltatory growth pattern. Tumor edema precedes peritumoral cyst development, which is often the harbinger of symptomatic presentation.
• Currently, surgical resection of hemangioblastomas in VHL patients is most often reserved for symptomatic lesions. In most cases, surgical removal of CNS hemangioblastomas can be performed safely and is effective and long-lasting.
• Resection of hemangioblastomas is guided towards the tumor, without resection, fenestration, shunting or other manipulation of the peritumoral cyst. The peritumoral cyst will resolve following resection of the hemangioblastoma.
• Endolymphatic sac tumors (ELSTs) may present as radiographic lesions in the petrous temporal bone of VHL patients, or clinically with hearing loss or other vestibulocochlear symptoms. Hearing loss may be sudden or insidious in onset. Hearing loss is a consequence of either tumor-related intralabyrinthine hemorrhage, tumor invasion of the otic capsule or through endolymphatic hydrops.
• Focused high-resolution radiographic investigation is warranted in a VHL patient with vestibulocochlear complaints.
• Surgical resection is performed on VHL patients with radiographically visible ELSTs to preserve hearing. Surgery may be considered in patients without radiographically evident ELSTs who have hearing loss and indirect evidence of an ELST, such as intralabyrinthine hemorrhage.
References
- Maher ER, Iselius L, Yates JR et al. von Hippel–Lindau disease: a genetic study. J. Med. Genet.28(7), 443–447 (1991).
- Maher ER, Yates JR, Harries R et al. Clinical features and natural history of von Hippel–Lindau disease. Q. J. Med.77(283), 1151–1163 (1990).
- Latif F, Tory K, Gnarra J et al. Identification of the von Hippel–Lindau disease tumor suppressor gene. Science260(5112), 1317–1320 (1993).
- Lonser RR, Glenn GM, Walther M et al. von Hippel–Lindau disease. Lancet361(9374), 2059–2067 (2003).
- Choyke PL, Glenn GM, Walther MM, Patronas NJ, Linehan WM, Zbar B. von Hippel–Lindau disease: genetic, clinical, and imaging features. Radiology194(3), 629–642 (1995).
- Karsdorp N, Elderson A, Wittebol-Post D et al. von Hippel–Lindau disease: new strategies in early detection and treatment. Am. J. Med.97(2), 158–168 (1994).
- Resche F, Moisan JP, Mantoura J et al. Haemangioblastoma, haemangioblastomatosis, and von Hippel–Lindau disease. Adv. Tech. Stand. Neurosurg.20, 197–304 (1993).
- Kaelin WG, Jr. The von Hippel–Lindau tumour suppressor protein: O2 sensing and cancer. Nat. Rev. Cancer8(11), 865–873 (2008).
- Walther MM, Reiter R, Keiser HR et al. Clinical and genetic characterization of pheochromocytoma in von Hippel–Lindau families: comparison with sporadic pheochromocytoma gives insight into natural history of pheochromocytoma. J. Urol.162(3 Pt 1), 659–664 (1999).
- Barontini M, Dahia PL. VHL disease. Best Pract. Res. Clin. Endocrinol. Metab.24(3), 401–413 (2010).
- Eisenhofer G, Lenders JW, Linehan WM, Walther MM, Goldstein DS, Keiser HR. Plasma normetanephrine and metanephrine for detecting pheochromocytoma in von Hippel–Lindau disease and multiple endocrine neoplasia type 2. N. Engl. J. Med.340(24), 1872–1879 (1999).
- Havekes B, King K, Lai EW, Romijn JA, Corssmit EP, Pacak K. New imaging approaches to phaeochromocytomas and paragangliomas. Clin. Endocrinol. (Oxf.)72(2), 137–145 (2010).
- Pacak K. Preoperative management of the pheochromocytoma patient. J. Clin. Endocrinol. Metab.92(11), 4069–4079 (2007).
- Walther MM, Choyke PL, Glenn G et al. Renal cancer in families with hereditary renal cancer: prospective analysis of a tumor size threshold for renal parenchymal sparing surgery. J. Urol161(5), 1475–1479 (1999).
- Joly D, Mejean A, Correas JM et al. Progress in nephron sparing therapy for renal cell carcinoma and von Hippel–Lindau disease. J. Urol185(6), 2056–2060 (2011).
- Neumann HP, Dinkel E, Brambs H et al. Pancreatic lesions in the von Hippel–Lindau syndrome. Gastroenterology101(2), 465–471 (1991).
- Hough DM, Stephens DH, Johnson CD, Binkovitz LA. Pancreatic lesions in von Hippel–Lindau disease: prevalence, clinical significance, and CT findings. Am. J. Roentgenol162(5), 1091–1094 (1994).
- Park DM, Zhuang Z, Chen L et al. von Hippel–Lindau disease-associated hemangioblastomas are derived from embryologic multipotent cells. PLoS Med.4(2), e60 (2007).
- Lonser RR, Vortmeyer AO, Butman JA et al. Edema is a precursor to central nervous system peritumoral cyst formation. Ann. Neurol.58(3), 392–399 (2005).
- Lonser RR, Butman JA, Oldfield EH. Pathogenesis of tumor-associated syringomyelia demonstrated by peritumoral contrast material leakage. Case illustration. J. Neurosurg. Spine4(5), 426 (2006).
- Wanebo JE, Lonser RR, Glenn GM, Oldfield EH. The natural history of hemangioblastomas of the central nervous system in patients with von Hippel–Lindau disease. J. Neurosurg.98(1), 82–94 (2003).
- Ammerman JM, Lonser RR, Dambrosia J, Butman JA, Oldfield EH. Long-term natural history of hemangioblastomas in patients with von Hippel–Lindau disease: implications for treatment. J. Neurosurg.105(2), 248–255 (2006).
- Takeuchi S, Tanaka R, Fujii Y, Abe H, Ito Y. Surgical treatment of hemangioblastomas with presurgical endovascular embolization. Neurol. Med. Chir (Tokyo)41(5), 246–251; discussion 251–242 (2001).
- Cornelius JF, Saint-Maurice JP, Bresson D, George B, Houdart E. Hemorrhage after particle embolization of hemangioblastomas: comparison of outcomes in spinal and cerebellar lesions. J. Neurosurg.106(6), 994–998 (2007).
- Horvathy DB, Hauck EF, Ogilvy CS, Hopkins LN, Levy EI, Siddiqui AH. Complete preoperative embolization of hemangioblastoma vessels with Onyx 18. J. Clin. Neurosci.18(3), 401–403 (2011).
- Jagannathan J, Lonser RR, Smith R, DeVroom HL, Oldfield EH. Surgical management of cerebellar hemangioblastomas in patients with von Hippel–Lindau disease. J. Neurosurg.108(2), 210–222 (2008).
- Lonser RR, Oldfield EH. Microsurgical resection of spinal cord hemangioblastomas. Neurosurgery57(4 Suppl.), 372–376; discussion 372–376 (2005).
- Mehta GU, Asthagiri AR, Bakhtian KD, Auh S, Oldfield EH, Lonser RR. Functional outcome after resection of spinal cord hemangioblastomas associated with von Hippel–Lindau disease. J. Neurosurg. Spine12(3), 233–242 (2010).
- Weil RJ, Lonser RR, DeVroom HL, Wanebo JE, Oldfield EH. Surgical management of brainstem hemangioblastomas in patients with von Hippel–Lindau disease. J. Neurosurg.98(1), 95–105 (2003).
- Wind JJ, Bakhtian KD, Sweet JA et al. Long-term outcome after resection of brainstem hemangioblastomas in von Hippel–Lindau disease. J. Neurosurg.114(5), 1312–1318 (2011).
- Filling-Katz MR, Choyke PL, Oldfield E et al. Central nervous system involvement in von Hippel–Lindau disease. Neurology41(1), 41–46 (1991).
- Neumann HP, Eggert HR, Weigel K, Friedburg H, Wiestler OD, Schollmeyer P. Hemangioblastomas of the central nervous system. A 10-year study with special reference to von Hippel–Lindau syndrome. J. Neurosurg.70(1), 24–30 (1989).
- Lonser RR, Wait SD, Butman JA et al. Surgical management of lumbosacral nerve root hemangioblastomas in von Hippel–Lindau syndrome. J. Neurosurg.99(1 Suppl), 64–69 (2003).
- Pluta RM, Wait SD, Butman JA et al. Sacral hemangioblastoma in a patient with von Hippel–Lindau disease. Case report and review of the literature. Neurosurg. Focus15(2), E11 (2003).
- Lonser RR, Butman JA, Kiringoda R, Song D, Oldfield EH. Pituitary stalk hemangioblastomas in von Hippel–Lindau disease. J. Neurosurg.110(2), 350–353 (2009).
- Peyre M, David P, Van Effenterre R et al. Natural history of supratentorial hemangioblastomas in von Hippel–Lindau disease. Neurosurgery67(3), 577–587; discussion 587 (2010).
- Chang SD, Meisel JA, Hancock SL, Martin DP, McManus M, Adler JR Jr. Treatment of hemangioblastomas in von Hippel–Lindau disease with linear accelerator-based radiosurgery. Neurosurgery43(1), 28–34; discussion 34–25 (1998).
- Asthagiri AR, Mehta GU, Zach L et al. Prospective evaluation of radiosurgery for hemangioblastomas in von Hippel–Lindau disease. Neuro Oncol.12(1), 80–86 (2010).
- Koh ES, Nichol A, Millar BA, Menard C, Pond G, Laperriere NJ. Role of fractionated external beam radiotherapy in hemangioblastoma of the central nervous system. Int. J. Radiat. Oncol. Biol. Phys.69(5), 1521–1526 (2007).
- Lonser RR, Baggenstos M, Kim HJ, Butman JA, Vortmeyer AO. The vestibular aqueduct: site of origin of endolymphatic sac tumors. J. Neurosurg.108(4), 751–756 (2008).
- Brandt R. Aur erage der angiomatosis retinae. Von Graefes Arch. Opthalmol.106, 127–136 (1921).
- Lindau A. [Studies on cerebellar cysts: Development, pathogenesis, and relationship with retinal angiomatosis]. Acta Path. Microbiol. Scand. Suppl.1, 1–128 (1926).
- Manski TJ, Heffner DK, Glenn GM et al. Endolymphatic sac tumors. A source of morbid hearing loss in von Hippel–Lindau disease. JAMA277(18), 1461–1466 (1997).
- Choo D, Shotland L, Mastroianni M et al. Endolymphatic sac tumors in von Hippel–Lindau disease. J. Neurosurg.100(3), 480–487 (2004).
- Megerian CA, Haynes DS, Poe DS, Choo DI, Keriakas TJ, Glasscock ME 3rd. Hearing preservation surgery for small endolymphatic sac tumors in patients with von Hippel–Lindau syndrome. Otol. Neurotol.23(3), 378–387 (2002).
- Kim HJ, Butman JA, Brewer C et al. Tumors of the endolymphatic sac in patients with von Hippel–Lindau disease: implications for their natural history, diagnosis, and treatment. J. Neurosurg.102(3), 503–512 (2005).
- Lonser RR, Kim HJ, Butman JA, Vortmeyer AO, Choo DI, Oldfield EH. Tumors of the endolymphatic sac in von Hippel–Lindau disease. N. Engl. J. Med.350(24), 2481–2486 (2004).
- Butman JA, Kim HJ, Baggenstos M et al. Mechanisms of morbid hearing loss associated with tumors of the endolymphatic sac in von Hippel–Lindau disease. JAMA298(1), 41–48 (2007).
- Jagannathan J, Butman JA, Lonser RR et al. Endolymphatic sac tumor demonstrated by intralabyrinthine hemorrhage. Case report. J. Neurosurg.107(2), 421–425 (2007).
- Mukherji SK, Albernaz VS, Lo WW et al. Papillary endolymphatic sac tumors: CT, MR imaging, and angiographic findings in 20 patients. Radiology202(3), 801–808 (1997).
- Richards PS, Clifton AG. Endolymphatic sac tumours. J. Laryngol. Otol.117(8), 666–669 (2003).
- Hashimoto M, Yokota A, Urasaki E, Imada H, Yamamoto H. Surgical treatment of endolymphatic sac tumor with adjunctive stereotactic radiation therapy – case report. Neurol. Med. Chir. (Tokyo)44(11), 595–599 (2004).
- Doherty JK, Yong M, Maceri D. Endolymphatic sac tumor: a report of 3 cases and discussion of management. Ear Nose Throat J.86(1), 30–35 (2007).
- Gupta R, Vaidhyswaran AN, Murali V, Kameswaran M. Endolymphatic sac papillary carcinoma treated with surgery and post-operative intensity-modulated radiotherapy: a rare case report. J. Cancer Res. Ther.6(4), 540–542 (2010).
- Patel PC, Pellitteri PK, Reams CL, Martin JS, Szymanski MB. Aggressive papillary tumor of the temporal bone: delayed extensive recurrence following radiation therapy. Skull Base Surg.7(1), 45–48 (1997).
- Lamiell JM, Salazar FG, Hsia YE. von Hippel–Lindau disease affecting 43 members of a single kindred. Medicine (Baltimore)68(1), 1–29 (1989).
- Melmon KL, Rosen SW. Lindau’s disease. Review of the literature and study of a large kindred. Am. J. Med.36, 595–617 (1964).
Management of von Hippel–Lindau disease-associated CNS lesions
To obtain credit, you should first read the journal article. After reading the article, you should be able to answer the following, related, multiple-choice questions. To complete the questions (with a minimum 70% passing score) and earn continuing medical education (CME) credit, please go to http://www.medscape.org/journal/expertneurothera. Credit cannot be obtained for tests completed on paper, although you may use the worksheet below to keep a record of your answers. You must be a registered user on Medscape.org. If you are not registered on Medscape.org, please click on the New Users: Free Registration link on the left hand side of the website to register. Only one answer is correct for each question. Once you successfully answer all post-test questions you will be able to view and/or print your certificate. For questions regarding the content of this activity, contact the accredited provider, [email protected]. For technical assistance, contact [email protected]. American Medical Association’s Physician’s Recognition Award (AMA PRA) credits are accepted in the US as evidence of participation in CME activities. For further information on this award, please refer to http://www.ama-assn.org/ama/pub/category/2922.html. The AMA has determined that physicians not licensed in the US who participate in this CME activity are eligible for AMA PRA Category 1 Credits™. Through agreements that the AMA has made with agencies in some countries, AMA PRA credit may be acceptable as evidence of participation in CME activities. If you are not licensed in the US, please complete the questions online, print the AMA PRA CME credit certificate and present it to your national medical association for review.
Activity Evaluation: Where 1 is strongly disagree and 5 is strongly agree
1. Your patient is a 35-year-old woman with von Hippel–Lindau (VHL) diagnosed with a cerebellar hemangioblastoma. Based on the review by Drs. Wind and Lonser, which of the following statements is most likely correct regarding considerations for surgery and perioperative management?
□ A All cerebellar hemangioblastomas should immediately be treated surgically
□ B She is unlikely to have any manifestations outside the central nervous system
□ C Management can be handled exclusively by the neurosurgical team
□ D Perioperative management needs may include pheochromocytomas, diabetes, and/or renal insufficiency
2. The patient described in question 1 has been having some gait instability but is generally in good health. Brain MRI shows a hemangioblastoma that appears to be surgically accessible and also shows a peritumoral cyst. Based on the review by Drs. Wind and Lonser, which of the following statements is most likely to apply to treatment approaches?
□ A Stereotactic radiosurgery is the best approach
□ B Preoperative embolization is indicated
□ C Resection should be guided toward the tumor alone
□ D The peritumoral cyst should be treated with fenestration or shunting
3. Based on the review by Drs. Wind and Lonser, which of the following statements about management of endolymphatic sac tumors (ELST) in patients with VHL is most likely correct?
□ A Radiation therapy is the best first-line therapy
□ B Patients with preserved hearing should be managed with a “wait-and-see” approach before considering surgery
□ C Surgical resection of ELST uses the retrolabyrinthine posterior petrosectomy approach
□ D Surgery on ELST should never be considered in patients who already have hearing loss
Notes
Data taken from Citation[4,56,57].