
Abstract
Direct cultivation-independent sequence retrieval of unidentified bacteria from histological tissue sections has been limited by the difficulty of selectively isolating specific bacteria from a complex environment. Here, a new DNA isolation approach is presented for prokaryotic cells. By this method, a potentially pathogenic strain of the genus Brachyspira from formalin-fixed human colonic biopsies were visualized by fluorescence in situ hybridization (FISH) with a 16S rRNA-targeting oligonucleotide probe, followed by laser capture microdissection (LCM) of the targeted cells. Direct 16S rRNA gene PCR was performed from the dissected microcolonies, and the subsequent DNA sequence analysis identified the dissected bacterial cells as belonging to the Brachyspira aalborgi cluster 1. The advantage of this technique is the ability to combine the histological recognition of the specific bacteria within the tissue with molecular analysis of 16S rRNA gene or other genes of interest. This method is widely applicable for the identification of noncultivable bacteria and their gene pool from formalin-fixed paraffin-embedded tissue samples.
Introduction
Histopathological diagnosis of bacterial infectious agents has traditionally been based on the use of formalin-fixed paraffin-embedded tissue samples. These samples represent a unique stock of archival disease-specific biological material. The identification of pathogenic microorganisms from formalin-fixed tissue samples has usually been done by PCR (Citation1–3). However, isolating high-quality genomic DNA from formalin-fixed tissue sections can be quite challenging. First, the fixation of tissue in formaldehyde leads to extensive protein-DNA cross-linking of all tissue components, and nucleic acids isolated from these specimens are highly fragmentated (Citation4). Secondly, there is a potential dilutional effect of the larger quantities of nontarget DNA found in whole tissue sections. The amplification product from formalin-fixed tissue can thus be reduced in quantity and size compared with fresh and frozen tissue (Citation5). Often histological tissue sections (e.g., from the gastrointestinal tract) represent a microbiologically complex environment, where previous knowledge of the microorganisms of interest is necessary in order to perform a specific PCR analysis. In this respect, the combination of histological recognition and the molecular identification and characterization of single cells or microcolonies of bacteria in tissue samples play an important role.
Laser capture microdissection (LCM) techniques have been used extensively in combination with analysis at the DNA and RNA level in eukaryotes (Citation6). Prokaryotes represent a different problem because of the smaller size of the cells and the lower amount of DNA compared to eukaryotic tissues. To our knowledge, only few other studies have previously used LCM and PCR for the isolation and identification of specific bacteria—and all in cases from species present in well-defined morphological areas that could be visualized by conventional histochemical staining methods such as hematoxylin and eosin (Citation7–10). However, with respect to bacteria, often tissue morphology alone is not sufficient for identification. This problem can be circumvented by fluorescence in situ hybridization (FISH), with ribosomal RNA (rRNA)-targeting oligonucleotide probes, which facilitates the rapid and specific identification of individual microbial cells within their natural environment (Citation11).
The present work describes the use of LCM combined with fluorescence in situ hybridization (LCM-FISH) to enable the phylogenetic analysis of a potentially pathogenic strain of Brachyspira dissected from formalin-fixed human colonic biopsies. Strains of Brachyspira aalborgi and Brachyspira pilosicoli have been reported to cause intestinal spiro-chetosis, which is visualized by a dense fringe of spirochetal bacteria attached to the colorectal epithelium forming a “false brush border” (Citation12,Citation13). The clinical significance of this colonization is disputed (Citation14), and knowledge about strain diversity and their pathogenic role is limited, mainly because members of the genus Brachyspira are fastidious microorganisms with specialized growth requirements. Different phylogenetic clusters of B. aalborgi have been identified (Citation15,Citation16), but the significance of these clusters in relation to clinical disease is unclear. The aim of this work was to develop the LCM-FISH technique and to identify Brachyspira cells in colon biopsy specimens.
Materials and methods
Samples
Three formalin-fixed paraffin-embedded biopsy specimens of the large intestine (colon) from three patients with a histopathologic diagnosis of human intestinal spirochetosis were obtained from S⊘rlandet Sykehus HF, Kristiansand, Norway. Three-micron-thick sections of the samples were mounted on 0.17-mm PALM® POL-membrane slides (P.A.L.M. Microlaser Technologies AG, Bernried, Germany) and kept at 4°C until use.
Hybridization of Formalin-Fixed Tissue Sections
Tissue sections were hybridized with the general bacterial probe, S-D-Bact-0338-a-A-18, or with a specific probe for the genus Brachyspira, L-G-Brachyspira-1410-a-A-19, 5′-labeled with Alexa Fluor® 488 or fluorescein isothio-cyanate, respectively (MWG-BIOTECH AG, Ebersberg, Germany) (). Prior to hybridization, tissue sections were deparaffinized with xylene and dehydrated in 96% ethanol for 30 min. Hybridization was carried out with 20 µL of hybridization buffer (100 mM Tris, pH 7.2, 0.9 M NaCl, 0.1% sodium dodecyl sulfate) and 100 ng of probe at 37°C for 16 h in a humidified chamber. The samples were washed in 100 mL of prewarmed (37°C) hybridization buffer for 15 min and subsequently in 100 mL of prewarmed (37°C) washing solution (100 mM Tris, pH 7.2, 0.9 M NaCl) for 15 min. The samples were finally rinsed in water and air-dried.
Table 1. Names and Sequences of 16S rRNA-Targeting Oligonucleotide Probes and 16S rRNA Gene Targeting Primers Used in This Study
Laser Capture Microdissection
A PALM Robot-microbeam system (P.A.L.M. Microlaser Technologies AG) consisting of an Axiovert 200 M microscope (Carl Zeiss, Oberkochen, Germany) equipped for epifluorescence with a 100-W Hg lamp, a 40×/1.30 oil Fluar objective (Carl Zeiss), filter set XF53 (Omega Optical, Brattleboro, VT, USA), and the PALM RoboSoftware version 1.2 (P.A.L.M. Microlaser Technologies AG) was used. Bacterial cells were selected by drawing around the area of interest and were dissected out of the tissue using the cutting and catapulting function, RoboLPC. The catapulted material was collected in the cap of a 200-µL Thermo-Tube (ABgene, Epsom, UK) containing 20 µL proteinase K buffer (see below).
DNA Extraction
The microdissected material was digested in proteinase K buffer (10 mM Tris-HCl, pH 8.0, 150 mM NaCl, 10 mM EDTA, 0.1% sodium dodecyl sulfate, 1 U proteinase K) at 55°C for 72 h. Subsequently, the proteinase K was inactivated at 95°C for 15 min. Two microliters of this solution was subsequently used as template for the PCR.
PCR and Sequencing
The primers (MWG-BIOTECH AG) used for 16S rRNA gene amplification are listed in . The following primer combinations were applied in order to amplify and partially sequence the 16S rRNA gene: primers 1 + 9 (457 bp), primers 2 + 10 (670 bp), primers 4 + 10 (325 bp), and primers 8 + 12 (383 bp). The following primer combinations were used in order to amplify and find maximum product length: primers 6 + 10 (324 bp), primers 7 + 11 (373 bp), primers 8 + 12 (383 bp), primers 1 + 9 (457 bp), primers 7 + 12 (490 bp), primers 5 + 10 (567 bp), primers 6 + 11 (662 bp), primers 3 + 10 (670 bp), and primers 6 + 12 (779 bp). The primers 1 + 3 (362 bp) were used in order to detect the minimum amount of tissue from which DNA could be amplified. The same primer combination was use to test if it was possible to obtain a PCR product of Brachyspira spp. from whole paraffin sections. Two paraffin sections for each patient were treated with proteinase K buffer (50 µL) for 72 h at 55°C and inactivated for 15 min at 96°C. Reaction conditions were as follows: 1×PCR Buffer II (Roche Applied Science, Indianapolis, IN, USA), 2 mM MgCl2, 400 µM of each deoxynucleotide triphosphate (Amersham Biosciences, Piscataway, NJ, USA), 0.4 µM of each primer, 0.5 U AmpliTaq® DNA Polymerase (Roche Applied Science), and 2.0 µL template DNA in a total volume of 50 µL. Thermal cycling using an MJ Research PTC-200® Thermal Cycler (Bio-Rad Laboratories, Hercules, CA, USA) was performed as follows: denaturation at 94°C for 3 min, followed by 35 cycles of denaturation at 94°C for 1 min, annealing at 55°C for 1 min, and extension at 72°C for 1.5 min, and then a final extension at 72°C for 10 min. Amplified PCR products were analyzed by electrophoresis in 1.5% agarose gels. The products were purified by using the QIAquick® spin PCR purification kit (Qiagen, Hilden, Germany) and sequenced by cycle sequencing on an ABI PRISM® 377 DNA Sequencer (Applied Biosystems, Foster City, CA, USA) using the ABI PRISM BigDye™ Terminator Cycle Sequencing kit (Applied Biosystems) according to the manufacturer’s instruction and the same primers as used for the PCR. Sequences were aligned to B. aalborgi NCTC11492T (GenBank® accession no. Z22781) and B. aalborgi W1 (GenBank accession no. AF200693) published 16S rRNA gene sequences using the ARB Sequence Editor v2.0 (www.arb-home.de).
Results and discussion
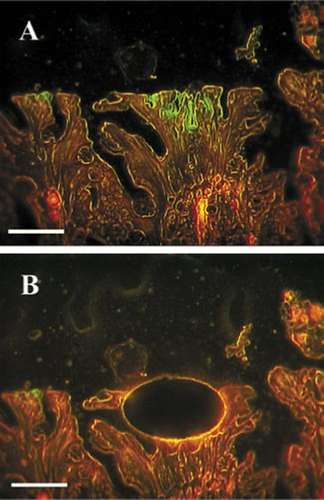
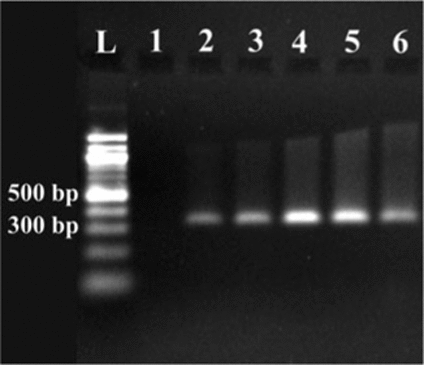
A Brachyspira strain from formalin-fixed paraffin-embedded human biopsy specimens of the colon was identified using an approach that incorporates FISH and the LCM technique in conjunction with 16S rRNA gene PCR and DNA sequence analysis. The three patients in this study all had a histopathologic diagnosis of human intestinal spirochetosis, which was confirmed by FISH analysis with a Brachyspira-specific 16S rRNA-targeting oligonucleotide probe (). For the LCM, the biopsy specimens were visualized with a bacterial 16S rRNA-targeting oligonucleotide probe (), which resulted in a clearly visible false brush border characteristic of a spirochete infection (). The green fluorescent microcolonies were microdissected with PALM microlaser technology () and collected in a cap of a PCR tube. In order not to lose any DNA, no extraction was performed. Instead, the PCR was done directly on 2 µL of the solution containing the microdissected tissue. The smallest amount of detectable bacterial DNA by PCR analysis was obtained from 4200 µm2 dissected tissue (). Consequently, by this method, it is possible to dissect and amplify DNA from small microcolonies. When PCR was done from whole paraffin sections, it was only possible to detect Brachyspira spp. from two of the three patients. This result is in concordance with previous results stating that better targeting of the source of DNA results in a more sensitive PCR (Citation7,Citation17).
The bacteria were targeted with a green fluorescent 16S ribosomal RNA (rRNA)-targeting oligonucleotide probe, while the intestinal cell material exhibited red autofluorescence. Scale bar, 250 µm. (A) Before microdissection. (B) After microdissection.
Areas of microdissected tissue from lane 1 to 6 were 1715, 4200, 7874, 37,936, 50,361, and 38,865 µm2, respectively. A 100-bp DNA ladder (L; New England Biolabs, Ipswich, MA, USA) was used.
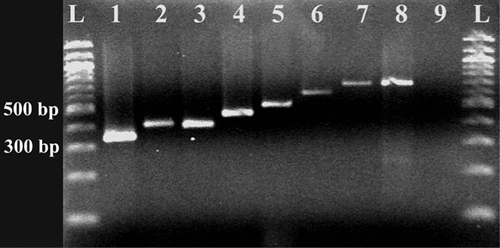
The effect of DNA fragmentation due to nucleic acid cross-linkage in formalin-fixated tissue was determined by performing PCR with primers amplifying nine different sizes of the 16S rRNA gene (). It was possible to reliably amplify fragments from 324 to 670 bp in size from the microdissected samples, while no 779-bp product was seen. This is in the range of previous results (Citation7,Citation9). Much larger fragments may have been amplified using other methods of fixation, such as frozen sections or methacarn fixation (Citation18,Citation19). But, in the case of the archival tissue used in the present investigation, this was not an option.
Expected sizes of products from lanes 1 to 9 were 324, 373, 438, 457, 490, 567, 662, 670, and 779 bp, respectively. A 100-bp DNA ladder (L) was used.
Using two different primer sets (1 + 3 and 2 + 4 in ) approximately 600 bp of the three specimens were PCR-amplified and sequenced. BLAST search in the National Center for Biotechnology Information (NCBI) database revealed a 99% nucleotide homology to B. aalborgi for all three sequences (results not shown). One of the isolates was further characterized by amplifying and sequencing 1435 bp of the 16S rRNA gene (GenBank accession no. AMO39526). This sequence had two and four nucleotide substitutions compared to B. aalborgi W1 and B. aalborgi NCTC11492T, respectively. Differences in nucleotide composition between the microdissected Brachyspira, henceforward referred to as NW719, and B. aalborgi W1 were as follows (positions given according to Escherichia coli numbering): Brachyspira NW719 had T and A in positions 209 and 630, while B. aalborgi W1 had C and G, respectively. The sequences for Brachyspira NW719 and B. aalborgi W1 had four extra guanosine residues in positions where B. aalborgi NCTC11492T lacks nucleotide information, however, the question is whether these are true differences or due to sequencing errors. Because of the high nucleotide similarity between the microdissected Brachyspira strain, B. aalborgiW1, and B. aalborgi NCTC11492T, we conclude that the microdissected strain belongs to the B. aalborgi cluster 1 (Citation16).
In conclusion, the LCM-FISH method presented here provides access to the genomic information and thus the identity of an unknown bacterium by combining histological recognition of the bacteria with molecular analysis of 16S rRNA gene. Furthermore, the identification by FISH allows the use of oligonucleotide probes targeting at different phylogenetic levels. In the present study, the bacterium under investigation had already been identified as a Brachyspira species, which allowed the use of primers specific for the genus Brachyspira. When no previous knowledge concerning the bacterium under investigation is available, universal 16S rRNA gene primers can instead be applied for the amplification and sequencing of approximately 500 bp. This method is widely applicable for the identification of unknown bacteria and their gene pool (e.g., noncultivable bacteria from archival formalin-fixed paraffin-embedded tissue samples or from complex environmental communities).
Competing Interests Statement
The authors declare no competing interests.
Acknowledgments
Kirstine Klitgaard and Lars M⊘lbak contributed equally to this work. We thank Katja Kristensen, Annie Ravn Pedersen, and Ulla Andreasen for excellent technical assistance. This work was supported by a grant from the Danish Agricultural and Veterinary Research Council project no. 23-02-0137 to Kirstine Klitgaard and a grant from The Ministry of Food, Agriculture, and Fisheries project no. 3401-65-03-745 to Lars M⊘lbak.
Additional information
Funding
References
- Bialek, R., A.Feucht, C.Aepinus, G.Just-Nubling, V.J.Robertson, J.Knobloch, and R.Hohle. 2002. Evaluation of two nested PCR assays for detection of Histoplasma capsulatum DNA in human tissue. J. Clin. Microbiol.40:1644–1647.
- Uzal, F.A., P.Hugenholtz, L.L.Blackall, S.Petray, S.Moss, R.A.Assis, M.M.Fernandez, and G.Carloni. 2003. PCR detection of Clostridium chauvoei in pure cultures and in formalin-fixed, paraffin-embedded tissues. Vet. Microbiol.91:239–248.
- Terio, K.A., L.Munson, L.Marker, B.M.Aldridge, and J.V.Solnick. 2005. Comparison of Helicobacter spp. in Cheetahs (Acinonyx jubatus) with and without gastritis. J. Clin. Microbiol.43:229–234.
- Wu, L., N.Patten, C.T.Yamashiro, and B.Chui. 2002. Extraction and amplification of DNA from formalin-fixed, paraffin-embedded tissues. Appl. Immunohistochem. Mol. Morphol.10:269–274.
- Greer, C.E., S.L.Peterson, N.B.Kiviat, and M.M.Manos. 1991. PCR amplification from paraffin-embedded tissues. Effects of fixative and fixation time. Am. J. Clin. Pathol.95:117–124.
- Simone, N.L., R.F.Bonner, J.W.Gillespie, M.R.Emmert-Buck, and L.A.Liotta. 1998. Laser-capture microdissection: opening the microscopic frontier to molecular analysis. Trends Genet.14:272–276.
- Ryan, P., M.W.Bennett, S.Aarons, G.Lee, J.K.Collins, G.C.Sullivan, J.Connell, and F.Shanahan. 2002. PCR detection of Mycobacterium paratuberculosis in Crohn’s disease granulomas isolated by laser capture microdissection. Gut51:665–670.
- Ryan, P., R.G.Kelly, G.Lee, J.K.Collins, G.C.O’Sullivan, J.O’Connell, and F.Shanahan. 2004. Bacterial DNA within granulomas of patients with Crohn’s disease-detection by laser capture microdissection and PCR. Am. J. Gastroenterol.99:1539–1543.
- Selva, E., V.Hofman, F.Berto, S.Musso, L.Castillo, J.Santini, P.Dellamonica, and P.Hofman. 2004. The value of polymerase chain reaction detection of Mycobacterium tuberculosis in granulomas isolated by laser capture microdissection. Pathology36:77–81.
- Yazdi, A.S., U.Puchta, M.J.Flaig, and C.A.Sander. 2004. Laser-capture microdissection: applications in routine molecular dermatopathology. J. Cutan. Pathol.31:465–470.
- Amann, R.I., W.Ludwig, and K.H.Schleifer. 1995. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol. Rev.59:143–169.
- Harland, W.A. and F.D.Lee. 1967. Intestinal spirochaetosis. BMJ3:718–719.
- Lee, J.I. and D.J.Hampson. 1994. Genetic characterisation of intestinal spirochaetes and their association with disease. J. Med. Microbiol.40:365–371.
- Munshi, M.A., N.M.Taylor, A.S.Mikosza, P.B.Spencer, and D.J.Hampson. 2003. Detection by PCR and isolation assays of the anaerobic intestinal spirochete Brachyspira aalborgi from the feces of captive nonhuman primates. J. Clin. Microbiol.41:1187–1191.
- Mikosza, A.S., M.A.Munshi, and D.J.Hampson. 2004. Analysis of genetic variation in Brachyspira aalborgi and related spirochaetes determined by partial sequencing of the 16S rRNA and NADH oxidase genes. J. Med. Microbiol.53:333–339.
- Pettersson, B., M.Wang, C.Fellstrom, M.Uhlen, G.Molin, B.Jeppsson, and S.Ahrne. 2000. Phylogenetic evidence for novel and genetically different intestinal spirochetes resembling Brachyspira aalborgi in the mucosa of the human colon as revealed by 16S rDNA analysis. Syst. Appl. Microbiol.23:355–363.
- Millar, D.S., S.J.Withey, M.L.Tizard, J.G.Ford, and J.Hermon-Taylor. 1995. Solid-phase hybridization capture of low-abundance target DNA sequences: application to the polymerase chain reaction detection of Mycobacterium paratuberculosis and Mycobacterium avium subsp. silvaticum. Anal. Biochem.226:325–330.
- Uneyama, C., M.Shibutani, N.Masutomi, H.Takagi, and M.Hirose. 2002. Methacarn fixation for genomic DNA analysis in microdissected, paraffin-embedded tissue specimens. J. Histochem. Cytochem.50:1237–1245.
- O’Leary, J.J., G.Browne, R.J.Landers, M.Crowley, I.B.Healy, J.T.Street, A.M.Pollock, J.Murphy, et al.. 1994. The importance of fixation procedures on DNA template and its suitability for solution-phase polymerase chain reaction and PCR in situ hybridization. Histochem. J.26:337–346.
- Alm, E.W., D.B.Oerther, N.Larsen, D.A.Stahl, and L.Raskin. 1996. The oligo-nucleotide probe database. Appl. Environ. Microbiol.62:3557–3559.
- Amann, R.I., L.Krumholz, and D.A.Stahl. 1990. Fluorescent-oligonucleotide probing of whole cells for determinative, phylogenetic, and environmental studies in microbiology. J. Bacteriol.172:762–770.
- Boye, M., T.K.Jensen, K.M⊘ller, T.D.Leser, and S.E.Jorsal. 1998. Specific detection of the genus Serpulina, S. hyodysenteriae, and S. pilosicoli in porcine intestines by fluorescent rRNA in situ hybridization. Mol. Cell. Probes12:323–330.
- Weisburg, W.G., S.M.Barns, D.A.Pelletier, and D.J.Lane. 1991. 16S ribosomal DNA amplification for phylogenetic study. J. Bacteriol.173:697–703.

