
Abstract
Confocal microscopes have evolved over the past 25 years from the early stage scanning systems to a collection of sophisticated laser scanning systems designed for a range of biomedical applications. Major improvements to the photon efficiency of the instrumentation coupled with the development of novel fluorescent reporters have enabled multidimensional imaging of living cells and tissues.
Looking Back
When BioTechniques made its debut in 1983, confocal microscopy had yet to become established in the world of biological imaging. Immunofluorescence microscopy was popular, and images of the distribution of fluorescent labels attached to antibodies to specific proteins were filling the journals with spectacular grayscale images (Citation1). The distribution patterns of many different proteins, especially cytoskeletal proteins, were giving novel views and insight into cellular structure and function at the level of the light microscope (Citation2). Around this time, fluorescent dyes were being developed that were sensitive to cellular physiology, such as calcium ion concentration (Citation3).
But there was a problem. The clearest images were collected from the thinnest regions of flattened cells growing on a glass coverslip in culture. The technique suffered from resolution issues when imaging even the thicker regions around the nucleus of cultured cells (). This was because the fluorescent signal from regions above and below the focal plain contributed to the images, and at best, blurred them, and often swamped out the signal completely. Most images of thicker specimens such as dividing cells, eggs, and embryos appeared as a bright fluorescent glow with little or no visible structure at all. Often structures of interest were visible in the light microscope but it was impossible to record them for publication. Researchers were forced to compromise by either physically cutting sections of their specimens or artificially flattening them in order to resolve structures of interest. Imaging living cells for structural and physiological studies was fraught with difficulty.
(A)–(C). Confocal imaging in the 1980s. (A) Wide-field epifluorescence image of a 3T3 cell immunofluorescently labeled with antitubulin. (B) Laser scanning confocal image of similar cell. (C) Double label confocal image of the same cell in (B); tubulin in green and nuclei labeled with propidium iodide in red. (D) Confocal imaging in the 1990s. Single optical sections collected simultaneously using a single krypton argon laser at three different excitation wavelengths—488 nm, 568 nm, and 647 nm—of a fruit fly third instar wing imaginal disk labeled for three genes involved with patterning the wing: cubitus interruptus (fluorescein, 496 nm) in green; vestigial (lissamine rhodamine, 572 nm) in red; and apterous (cyanine 5, 649 nm) in blue. (E) Confocal imaging in the 21st century. Brainbow image of mouse dentate gyrus (Citation21).
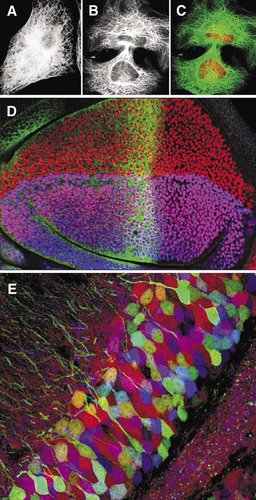
There was a desperate need to improve the resolution of images collected using fluorescence microscopy. One possibility was confocal microscopy. The technique had been around for many years before 1983: in 1957 Marvin Minsky had patented an instrument designed for studying neural networks in the living brain, but it was not sensitive enough for fluorescence and was not well-suited to biological specimens since the stage rather than the light beam moved, causing vibration artifacts (Citation4,Citation5). In 1983, however, the technology was not sufficiently well-advanced to produce a confocal instrument suitable for collecting images of fluorescently labeled specimens on a routine basis. But it was close.
Coming of Age
The practical application of confocal microscopy for biological fluorescence imaging was introduced some four years later in 1987 with the introduction of the first commercially available laser scanning confocal microscopes (Citation6,Citation7,Citation8). The approach was typified by the MRC 500 laser scanning confocal microscope (Bio-Rad Microsciences, Hemel Hempstead, England), which was designed to interface with an existing epifluorescence microscope rather like a video camera. The instrument consisted of a scanning unit that contained the electronics to drive two mirrors to produce a focused scanning beam in the specimen from an air-cooled argon laser together with photomultiplier tube detectors (PMT). The output from the PMTs was built into an image using a framestore card in a microcomputer, and the resulting digital image was displayed on the screen of the computer at a rate of about one full 768 × 512 pixel frame per second. Hard copies of the image files were collected by photographing the screen of the video monitor and archived digitally.
The images obtained using the instrument were a strong demonstration of the power of the confocal approach for fluorescently labeled specimens. Optical sections that showed subcellular details from within thick and brightly labeled specimens were obtained for the first time from a range of fixed cells and tissues, and with little additional specimen preparation as compared with that required for conventional epifluorescence microscopy. The liberal application of antibleaching agents, however, was highly recommended.
A powerful “proof of performance” of the laser scanning confocal approach convinced a large section of cell biologists of its value for imaging immuno-fluorescently labeled specimens (Citation7). This publication included a veritable who’s who of images of specimen types from major areas of contemporary biomedical research. The specimens included unflattened HeLa cells, mitotic spindles, fertilized sea urchin eggs, spherical plasmacytoma cells, salivary gland preparations of Drosophila larvae, C. elegans embryos, nerve tissues, and chick embryos. In addition to single optical sections (), the instrument could collect serial optical sections for later 3-D reconstructions, profiles of cells (x-z sections), and reflected light images. The power of the confocal approach lies in the ability to image structures at discrete levels within an intact biological specimen with a theoretical lateral resolution of 0.14 µm and a vertical resolution of 0.23 µm using a lens of 1.4 numerical aperture. These first instruments could also collect images from specimens labeled with two fluorochromes () as well as transmission images. The images were important for mapping the distribution of the protein of interest onto landmarks within cells and tissues.
Every Photon is Sacred
As the numbers of users increased, the experimental demands on the early instruments were pushed to the limits. Many different experimental approaches were now theoretically viable options with the ability to scan a specimen with a laser beam under the control of a microcomputer, as with fluorescence recovery after photobleaching, for example. The early instruments, however, were relatively wasteful of photons, which meant they were fine for imaging brightly labeled fixed specimens where photobleaching or photodamage to the cells was not an issue. Such a wasteful light budget made imaging living cells and tissues a difficult option since observations were often compromised by worries concerning artifacts from photodamage. Moreover, multiple label images often suffered from bleed-through from one channel to another and photobleaching of probes because of the high laser powers required for excitation of the fluorochromes, which made definitive analysis of relative distributions of proteins problematic.
The instruments available today are vastly different from the early models available in 1987. They have evolved over years of incorporation of technological advancements from multiple sources into all parts of the imaging chain from the lasers, fiber optic couplings, electronic filters, scanning mechanisms, spectral detectors, and also in computer hardware, software, and improved methods of digital display and reproduction of images.
The “optical section” is now an accepted part of the cell biologist’s vernacular, and the spectacular images from these instruments adorn the journals and web sites of the world. There are now many different instruments available for collecting optical sections in addition to the laser scanning confocal approach (Citation9). Instruments have been developed for more specific applications. For example, multiple photon microscopes allow imaging more deeply into tissues and reduce photodamage to fluorophores and tissues when compared with the laser scanning approach (Citation10). Multiple beam scanning systems and digital deconvolution of wide-field images are also practical options for collecting optical sections.
Several studies have compared the performance of confocal, multiple photon, and wide-field deconvolution microscopy on a variety of fluorescently labeled specimens (Citation11,Citation12). Wide-field deconvolution is preferred for thin and relatively dim samples, for example yeast cells, whereas the confocal and multiple photon imaging are preferred for imaging thick and bright samples, such as whole mounts of tissues and embryos and for imaging in vivo. The multiple photon approach is preferred to confocal microscopy for imaging deeper into living tissues. The multiple beam scanning confocal microscopes are preferred for imaging faster events at full resolution whereas the laser scanning confocal microscopes can be used for measuring fast events at low resolution.
The different kinds of data acquired from confocal microscopes have been classed as “multidimensional.” The number of dimensions has now reached five: the X, the Y, and the Z planes, together with time as the fourth dimension and wavelength as the fifth dimension. Thus five-dimensional imaging would be a time-lapse series of Z-series of multichannel images (Citation13). It is clear that datasets acquired from multidimensional experiments can be relatively large and present problems with data management. Concerted efforts are under way to standardize datasets from different imaging systems across the world (Citation14).
The Fifth Dimension
Perhaps one of the most spectacular developments in confocal imaging is the ability to collect multiple images in the fifth dimension—that of wavelength. From the early days, one of the major applications of the laser scanning confocal microscope was for multiple label imaging, since the registration between the images collected with the same scanning beam at different excitation wavelengths was usually maintained assuming the cells did not move and a sufficiently well-corrected objective lens was used. Two-color imaging using fluorescein and rhodamine probes was quickly achieved, along with the ability to merge a nonconfocal transmitted light image with the fluorescence images.
In the early 1990s three-channel imaging () became a routine technique using Krypton-Argon gas laser sources (or combinations of lasers) and a scanning head equipped with three PMTs that were able to collect the three channels simultaneously (Citation15). Around this time the green fluorescent protein (GFP) was isolated from the jellyfish Aequorea victoria, and was used in Caenorhabditis elegans as a genetically stable fluorescent marker that could be imaged in the confocal microscope at the same excitation wavelength (around 488 nm) as fluorescein in living animals. Fluorescent proteins such as GFP and others have been mutated to give reporters of different spectral properties so that multicolor labeling in living animals is now a practical option (Citation16–18). Such probes can be used to follow physiological processes within living cells using fluorescence resonance energy transfer (Citation19) or probes for different stages of the cell cycle (Citation20).
Over the past few years there have been improvements made for imaging more than three probes in the same sample, and developments now allow the detection of multiple channels. This has given a finer control of bleed-through between channels, improved rejection of autofluorescence, and the ability to separate probes whose emission spectra overlap. The current generation of confocal microscopes has been used to stunning effect to produce spectacular images of genetically labeled neurons in combinations of three or more fluorescent proteins (Citation21). Using the Brainbow technique, an unprecedented 90 different colors of labeled neurons were detected in a single sample using a standard laser scanning confocal microscope equipped with a spectral detector (Olympus FV1000, Olympus America, Melville, NY, USA) (). Moreover, these probes are so stable and brightly expressed in vivo that they enable multicolor imaging of living neurons.
Beyond the Rainbow
Confocal microscopy continues to find applications in all branches of biological imaging for routine analysis, including pathology. Major developments are being made in high-resolution live cell imaging and for imaging in vivo (Citation22,Citation23). Early confocal instruments were able to detect two or three fluorochromes. The number of different colors in the current generation of instruments stands at 90. Is there room for more channels? Perhaps, considering there are 13,659 protein-coding genes in the Drosophila genome (Citation24,Citation25)!
Competing Interests Statement
The author declares no competing interests.
Acknowledgments
I thank Jim Williams for preparing the specimen for . Jean Livet, Tamily Weissman, and Jeff Lichtman, Dept. of Molecular and Cellular Biology and Center for Brain Science at Harvard University kindly supplied the Brainbow image, . Finally, I would like to thank Sean Carroll for our continued collaboration.
Additional information
Funding
References
- Lazarides, E. and K.Weber. 1974. Actin antibody: the specific visualization of actin filaments in non-muscle cells. Proc. Natl. Acad. Sci. USA71:2268–2272.
- Goldman, R.D., T.Pollard, and J.Rosenbaum (Eds.). 1976. Cell Motility (Cold Spring Harbor Conferences on Cell Proliferation), p. 1373. Books A, B, C. Vol. 3. CSH Laboratory Press, Cold Spring Harbor, NY.
- Tsien, R.Y., T.J.Rink, and M.Poenie. 1985. Measurements of cytosolic free calcium individual small cells using fluorescence microscopy with dual excitation wavelengths. Cell Calcium6:145–157.
- Minsky, M. 1988. Memoir on inventing the confocal scanning microscope. Scanning10:128–138.
- Minsky, M. 1957. Microscopy apparatus. U.S. Patent 3013467.
- White, J.G. and W.B.Amos. 1987. Confocal mi croscopy comes of age. Nature328:183–184.
- White, J.G, W.B.Amos, and M.Fordham. 1987. An evaluation of confocal versus conventional imaging of biological structures by fluorescence light microscopy. J. Cell Biol.105:41–48.
- Amos, W.B. and J.G.White. 2003. How the confocal laser scanning microscope entered biological research. Biol. Cell95:335–342.
- Pawley, J.B. (Ed.). 2006. Handbook of Biological Confocal Microscopy, 3rd ed.Springer, New York.
- Svoboda, K. and R.Yasuda. 2006. Principles of two-photon excitation microscopy and its applica tions to neuroscience. Neuron50:823–839.
- Swedlow, J.R., K.Hu, P.D.Andrews, D.S.Roos, and J.M.Murray. 2002. Measuring tubulin content in Toxoplasma gondii: a comparison of laser-scanning confocal and wide-field fluorescence micros copy. Proc. Natl. Acad. Sci. USA99:2014–2019.
- Periasamy, A., P.Skoglund, C.Noakes, and R.Keller. 1999. An evaluation of two-photon excitation versus confocal and digital deconvolution fluores cence microscopy imaging in Xenopus morphogenesis. Microsc. Res. Tech.47:172–181.
- Andrews, P.D., I.S.Harper, and J.R.Swedlow. 2002. To 5D and beyond: quantitative fluorescence microscopy in the postgenomic era. Traffic3:29–36.
- Swedlow, J.R., I.Goldberg, E.Brauner, and P.K.Sorger. 2003. Informatics and quantitative analysis in biological imaging. Science300:100–102.
- Brelje, T.C., M.W.Wessendorf, and R.L.Sorenson. 1993. Multicolor laser scanning confocal immunofluorescence microscopy: practical applications and limitations. Methods Cell Biol.38:98–181.
- Lichtman, J.W. and S.E.Fraser. 2001. The neuronal naturalist: watching neurons in their native habitat. Nat. Neurosci.4:1215–1220.
- Jacobs, R.E., E.T.Ahrens, T.J.Meade, and S.E.Fraser. 1999. Looking deeper into vertebrate development. Trends Cell Biol.9:73–76.
- Hadjantonakis, A.K., M.E.Dickinson, S.E.Fraser, and V.E.Papaioannou. 2003. Technicolour transgenics: imaging tools for functional genomics in the mouse. Nat. Rev. Genet.4:613–625.
- Piston, D.W. and G.J.Kremers. 2007. Fluorescent protein FRET: the good, the bad and the ugly. Trends Biochem. Sci.32:407–414.
- Sakaue-Sawano, A., H.Kurokawa, T.Morimura, A.Hanyu, H.Hama, H.Osawa, S.Kashiwagi, K.Fukami, T.Miyata, et al.. 2008. Visualizing spatio-temporal dynamics of multicellular cell-cycle pro gression. Cell132:487–498.
- Livet, J., T.A.Weissman, H.Kang, R.W.Draft, J.Lu, R.A.Bennis, J.R.Sanes, and J.W.Lichtman. 2007. Transgenic strategies for combinatorial expression of fluorescent proteins in the nervous system. Nature450:56–62.
- Huang, B., W.Wang, M.Bates, and X.Zhuang. 2008. Three-dimensional super-resolution imaging by stochastic optical reconstruction microscopy. 2008. Science319:810–813.
- Holtmaat, A., L.Wilbrecht, G.W.Knott, E.Welker, and K.Svoboda. 2006. Experience-dependent and cell-type-specific spine growth in the neo-cortex. Nature441:979–983.
- Tomancak, P., B.P.Berman, A.Beaton, R.Weiszmann, E.Kwan, V.Hartenstein, S.E.Celniker, and G.M.Rubin. 2007. Global analysis of gene expression during Drosophila embryogenesis. Genome Biol.8:R145.
- Li, X., S.MacArthur, R.Bourgon, D.Nix, D.Pollard, V. N.Iyer, A.Hechmer, L.Simirenko, et al.. 2008. Transcription factors bind thousands of active and inactive regions in the Drosophila blastoderm. PLOS Biology6:365–388.

