
Abstract
Genetic tools are required to take full advantage of the wealth of information generated by genome sequencing efforts and ensuing global gene and protein expression analyses. Bacterial genetics was originally developed and refined in Escherichia coli. As a consequence, elegant plasmid, cloning, expression, and mutagenesis systems were developed over the years and a good number of them are commercially available. This is not true for other bacteria. Although the development of genetic tools has generally not kept up with the sequencing pace, substantial progress has been made in this arena with many bacterial species. This short review highlights selected topics and achievements in the field over the past 25 years and presents some strategies that may help address future challenges. BioTechniques has played an integral part in the publication of important technological advances in the field over the first 25 years of its existence and it can be anticipated that it will continue to do so in the future.
Introduction
One may argue that among the technologies that had the greatest impact on the field of bacterial genetics in the last 25 years is the polymerase chain reaction (PCR) (Citation1,Citation2), for its effect on strategies for cloning and manipulation of genes (Citation3–5). However, PCR is undoubtedly rivaled by the emergence of whole-genome shotgun sequencing technologies (Citation6) that, to date, have allowed deciphering of several hundred bacterial genome sequences. Besides postgenomic methods such as transcriptome, proteome, and metabolome analyses, genetic tools are needed to take advantage of the wealth of information contained within these genomes. However, with the notable exception of PCR-based strategies, tools for genetic analysis have largely not kept up with the “sequencing pace.” Many bacteria are recalcitrant to genetic manipulation because of the lack of suitable selection markers, necessitating either identification of novel selection markers or use of markers that can be repeatedly used (“recycled”). Furthermore, tools for genetic manipulation are either scarce or nonexistent, or they exhibit a narrow host-range. This may be remedied by developing tools that are as broad-host-range as possible. The new technologies needed may not necessitate de novo identification of elements and tools that work in the bacterium of choice, but in most instances, can probably be derived by appropriate modification of what is already available for other bacteria. In this short paper, I review some of the technologies that have emerged over the last 25 years and how they may help meet the challenges the future of bacterial genetics holds. Because of space limitations, this review cannot be all-encompassing, but I hope that it will nonetheless be thought-provoking to the reader and encourage further searching and reading of the pertinent literature.
Cloning Vectors
Plasmid cloning vectors for Escherichia coli were developed many years ago. Most rely on the pMB1 (closely related to ColE1), p15A, or pSC101 replicons. Examples for pMB1-based vectors include the pBR series of vectors of intermediate copy number (15–20 per cell) that contain various antibiotic resistance markers for cloning via insertional inactivation (reviewed in Reference Citation7). Subsequent modifications by Messing and colleagues created the pUC series of vectors with an increased plasmid-copy number (>500 copies per cell) and facilitated cloning into a multiple cloning site (MCS) (Citation8,Citation9). This MCS is located within the lacZα gene segment of E. coli β-galactosidase (β-Gal) (Citation8) and allows blue-white screening in appropriate host strains expressing the LacZΔM15 β-Gal protein (Citation10). Other versions of similar high-copy number vectors are exemplified by the widely used pBluescript vectors (Citation11). The pACYC vectors were the first examples of p15A-based vectors of lower copy number (∼10 copies per cell), which were also engineered for cloning by insertional inactivation of ampicillin, chloramphenicol, kanamycin, and tetracycline resistance markers (Citation12). Later derivatives containing the p15A replicon included vectors that carry the pUC-derived MCSs and β-Gal-based blue-white screening elements (Citation13,Citation14). Lastly, pSC101 was one of the very first low-copy number (∼5 copies per cell) plasmids engineered for cloning of heterologous DNA fragments (Citation15). The pSC101 replicon was later combined with the pBluescript selection markers and MCS to derive versatile low-copy number cloning vectors (Citation16).
Some of these vectors were later modified to allow cloning of PCR fragments via TA (Citation17) or Vaccinia DNA topoisomerase I based TOPO (Citation18) cloning, which are commercially marketed technologies. Additionally, the commercially available Gateway system (Invitrogen, Carlsbad, CA, USA) allows rapid recombination-based cloning of DNA fragments into recombination-proficient, so-called destination vectors, by exploiting the efficient phage λ recombination machinery (Citation19).
The ColE1, p15A, and pSC101 replicons only function in E. coli and some other closely related Enterobacteriaceae. There are basically two strategies for development of plasmid cloning vectors for bacteria other than E. coli: (i) one can search for cryptic plasmids present in these bacteria and exploit their replicons for development of cloning vectors (Citation20,Citation21) or (ii) one can exploit vectors that have been shown to be of broad-host-range such as pBBR1 (Citation22), pRO1600 (Citation20), RK2 (Citation23), RSF1010 (Citation24), or pVS1 (Citation25). Because of the time-consuming nature of the former approach, the latter is more feasible and has been exploited by several groups for development of broad-host-range vectors using strategies similar to those illustrated in . These efforts resulted in small and well-characterized pBBR1-based vectors such as pBBR1MCS (Citation26,Citation27) or the pRO1600-based pUCP family of vectors (Citation28–31). More comprehensive reviews of these and other replicon-based cloning vectors have been published (Citation32,Citation33). Because of the ease of manipulation of E. coli and the many reagents available for this bacterium, most cloning vectors available for other bacterial hosts are shuttle plasmids that replicate in both E. coli and other bacteria (e.g., contain a pMB1-derived replicon and the broad-host-range replicon). Besides a replicon for stable maintenance in the respective hosts, other basic requirements include a selection marker for identification of cells containing and maintaining the plasmid and restriction sites (e.g., a MCS) for cloning of DNA fragments. While not essential, other features are often included to increase the versatility of the vectors. These include genetic elements facilitating screening of recombinants (e.g., lacZα for blue-white screening); elements that facilitate interspecies transfer of recombinant plasmids (e.g., an origin of conjugal transfer, mob or oriT); promoters whose expression levels can be regulated (e.g., E. coli lac operon promoter Plac or its derivatives Ptac and Ptrc (Citation34)); other E. coli promoters such as those derived from the arabinose (ParaBAD) (Citation35,Citation36) and rhamnose (PrhaBAD) (Citation37) operons; tags (e.g., oligohistidine (Citation38–40)) that facilitate purification of recombinant proteins.
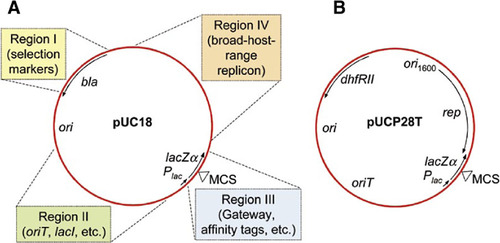
(A) The narrow-host-range vector pUC18 possesses several desirable features of a versatile cloning vector: (Citation1) it is small (2686 bp) and its entire nucleotide sequence is known; (Citation2) it contains an ampicillin resistance selection marker encoded by the β-lactamase (bla) gene; (Citation3) it contains a pMB1-derived high-copy number origin of replication; (Citation4) it contains a multiple cloning site (MCS) for versatile fragment cloning; (Citation5) it contains the E. coli β-galactosidase α fragment-encoding gene (lacZα), which allows blue white-screening in hosts expressing the LacZΔM15 protein; and (Citation6) it contains the Escherichia coli lac operon promoter (Plac), which allows regulated expression of cloned genes in hosts expressing the lacI-encoded Lac repressor or constitutive expression of cloned genes in hosts devoid of lacI. Despite its small size, pUC18 has at least four dispensable regions that can be exploited for broad-host-range vector development and/or insertion of other functional elements, some of which are indicated in the colored boxes (e.g., an origin of transfer, oriT; the Lac repressor-encoding lacI gene and others). (B) The broad-host-range vector pUCP28T was derived by inserting functional elements into three of the four dispensable regions of pUC18: (Citation1) the bla gene was deleted and replaced with the trimethoprim resistance-encoding dhfRII gene; (Citation2) the oriT was inserted into region II; and (Citation3) a cassette containing the pRO1600 broad-host-range origin of replication (ori1600) and its associated replication protein gene (rep) was inserted into region IV. Despite gain of function by insertion of various functional elements, the resulting mobilizable shuttle vector still maintains several of the features found in pUC18, that is, small size (4080 bp), versatile MCS, blue-white selection, and expression of cloned inserts from Plac. Similar strategies can be used for conversion of other narrow-host-range vectors into broad-host-range vectors.
Alternatives to autonomously replicating plasmid vectors are broad-host-range single-copy chromosomal integration systems such as those typified by Tn5-based vectors (random integration) (Citation41,Citation42) and Tn7-based vectors (site-specific integration) (Citation43,Citation44). In contrast to random insertion in the chromosome, which has the propensity to result in undesired mutants, the latter has the obvious advantage that integration occurs with high frequency at a neutral, naturally evolved site (attTn7) found downstream of the essential glmS gene in many bacteria (Citation45). Insertion site specificity is not provided by attTn7, but by sequences located within the essential glmS gene and the TnsD protein of the Tn7 transposase complex. Insertion at sites other than those situated downstream of glmS is rare and has only been observed in Proteus mirabilis where, besides a glmS-linked site, a secondary attTn7 site has been identified within the carAB operon (Citation46). It can be assumed that chromosomes with glmS probably possess at least one attTn7 site. In fact, it has been shown that some bacteria possess multiple glmS genes and thus multiple attTn7 sites (Citation44,Citation47,Citation48). Owing to minimal system requirements—left (Tn7L) and right (Tn7R) Tn7 transposon ends and the site-specific transposase complex in trans (e.g., TnsABCD)—mini-Tn7 elements have been engineered that incorporate all features traditionally found in plasmids, for example, antibiotic selection markers and regulatable promoters. These elements are applicable in a wide range of bacteria. In contrast to most plasmid vectors that necessitate continuous selection, Tn7-based vectors replicate stably along with the chromosome and thus are suitable for use in environments where antibiotic selection is not feasible, such as animal and plant models, biofilms, and bacteria destined for environmental release. Despite their widespread utility in Gram-negative bacteria, Tn7-derived vectors have not yet been successfully applied in Grampositive bacteria.
Selection Markers
Once established in the respective host(s), many plasmids are stably maintained. However, selection markers are required for selection of plasmid-containing colonies after transformation or conjugation experiments and to ensure that each cell in a bacterial population contains a plasmid. Useful selection markers include, in order of decreasing importance, those conferring resistance to antibiotics, other antimicrobials, heavy metals, and metabolic markers.Numerous plasmids have been developed that serve as sources for various antibiotic resistance cassettes (Citation49–54). The choice of antibiotic selection marker(s) depends largely on the susceptibility of the host, and intrinsic resistance to many antibiotics quite often hampers use of plasmids in bacteria. Intrinsic resistance or regulatory restrictions (e.g., those encountered with Select Agent bacteria in the United States) quite often leave only a few useful and approved antibiotic selection markers. In these instances, one can consider implementation of broad-host-range site-specific recombinase systems that allow recycling of the precious few markers remaining for some bacteria. Removal of antibiotic resistance markers is often also required during construction of genetically modified organisms in the food industry. The two most commonly used systems are the Flp/FRT system from Saccharomyces cerevisiae (Citation55,Citation56) and the Cre/loxP system from bacteriophage P1 (Citation57–59), for which broad-host-range versions have been engineered (Citation60–62).
Nonantibiotic resistance markers (e.g., triclosan (Citation63) or heavy metals such as tellurite (Citation64)) are attractive for bacteria in situations when the use of antibiotic resistance markers is restricted (e.g., Select Agent bacteria in the United States) or in bacteria destined for environmental release. As with antibiotics, marker choice depends on susceptibility of the bacterial host. In contrast to the many antibiotic resistance markers available, only a few nonantibiotic resistance marker-encoding gene cassettes have been described. This, however, may change in light of recently implemented National Institutes of Health (NIH) funding initiatives aimed at developing nonantibiotic markers to facilitate research with Select Agent bacteria in the United States.
Metabolic markers are most useful in environments where selection with antimicrobials is less feasible, for instance, in bacteria destined for environmental release. Because many bacteria lack the required genes to metabolize lactose, the E. coli lactose operon can be used as a selectable marker in these bacteria (Citation65). The use of other metabolic markers is usually not feasible in wild-type bacteria because they require host mutations that allow for selection of the plasmid-encoded markers.
Counter-selection Markers
In contrast to selection markers, counter-selection markers serve to eliminate unwanted elements. These elements can be helper strains used during conjugation experiments or DNA segments introduced during genetic manipulation experiments, for example, as with transposon delivery.
Counter-selection against bacteria can be achieved by exploiting intrinsic properties present in one bacterium but not another. These include intrinsic resistance to antimicrobials or unique metabolic properties. For example, in contrast to E. coli, Pseudomonas aeruginosa is resistant to triclosan (also known as irgasan) and can use citrate as a sole carbon and energy source. Consequently, both triclosan-containing agar (e.g., Pseudomonas isolation agar from Difco, Detroit, MI, USA) as well as citrate minimal media (e.g., Vogel-Bonner minimal medium (Citation66)) can be used to counter-select against E. coli in conjugation experiments. Similarly, Burkholderia spp. are intrinsically resistant to cationic peptides, which allows the use of agar containing polymyxin B to counter-select against E. coli (Citation47).
Several counter-selection methods can be applied to eliminate unwanted DNA sequences. Plasmids containing temperature-sensitive (Ts) replicons are maintained at permissive temperatures (usually 30°C) and eliminated at temperatures >37°C (Citation67–70). In many instances, such Ts replicons are easily obtained by error-prone PCR (Citation48,Citation71). Other methods involve incorporation of sequences that encode enzymes whose expression is toxic in the presence of certain substrates. Some examples include the following: (i) sequences containing the sacB gene from Bacillus subtilis confer sucrose-sensitivity when expressed in E. coli and other Gram-negative bacteria. Selection of sucrose-resistant derivatives eliminates sacB-containing sequences (Citation72–78); (ii) the wild-type rpsL gene confers streptomycin susceptibility in a rpsL mutant background. This marker can therefore also be used to counter-select against unwanted DNA sequences in any background in which streptomycin resistance can be selected (Citation79,Citation80); (iii) the bacterial pheS gene encodes a phenylalanyl-t-RNA synthetase α subunit. Mutation of G294A in this protein results in an enzyme with relaxed substrate specificity that tolerates incorporation of the toxic phenylalanine analog p-chlorophenylalanine. Expression, therefore, of a mutated pheS gene in the presence of p-chlorophenylalanine is toxic to the host cell (Citation81). This strategy has successfully been used for counter-selection in Gram-negative (Citation81–83) and Gram-positive bacteria (Citation84). Although for maximal efficiency in the chosen host, it may be necessary to clone the endogenous pheS gene and engineer the E. coli-homologous allele to incorporate the G294A substitution. There are alternative counter-selection procedures involving toxic compounds that have been less widely used. One involves the use of the amiE gene of P. aeruginosa, the product of which (an aliphatic amidase) converts fluoroacetamide to the toxic compound fluoroacetate (Citation85). Another exploits the finding that expression of the S. cerevisiae URA3 gene in an E. coli pyrF mutant in the presence of 5-fluoroacetic acid (5-FOA) leads to its metabolism into a suicide substrate (Citation86). Lastly, the E. coli toxin gene mazF was used for generation of marker-less mutations in Bacillus subtilis (Citation87). Other counter-selection markers have been described and reviewed (Citation88), but most of them were developed for use with E. coli and may have limited applicability outside of the Enterobacteriaceae.
Choice of the counter-selection marker(s) depends largely on the physiology of the host bacterium. For example, the sacB- and rpsL-based counter-selection strategies are not applicable in wild-type Burkholderia spp. because they contain endogenous sacB (Citation89) genes and are intrinsically streptomycin-resistant (Citation90). In these bacteria, pheS-based counter-selection is currently the only viable method.
Transformation Procedures
Genetic manipulation of bacteria requires transfer of recombinant DNA molecules, either circular plasmids or linear DNA fragments, into the intended host(s). The four basic methods used for transfer of DNA molecules into bacteria are: (i) natural transformation; (ii) transformation using chemical competent cells; (iii) electroporation into electrocompetent cells; and (iv) conjugation.
Natural transformation is relatively common in bacteria (Citation91). The methods available for isolating chemically competent cells are too numerous to be reviewed here (a Google search with the terms “chemical competent cells” at the time of writing of this review yielded well over 150,000 hits). Chemically prepared competent cells are available from many commercial sources and can also easily be prepared and stored. Many methods for preparation of electrocompetent cells and electroporation have either been published or are available online (at the time of writing of this review, a Google search with the terms “electrocompetent cells” yielded about 40,000 returns). In general, electroporation methods can be more readily adapted than chemical transformation methods for use in bacteria other than E. coli.
While transformation methods are generally simple and efficient, conjugation is still a widely used and often more efficient DNA transfer method, especially for larger plasmids. Because the transfer regions contained on these plasmids, such as the mob region from RSF1010 or the oriT from RK2, are minimal, they are incapable of self-transfer and rely on mobilization functions provided in trans. To facilitate construction of mobilizable plasmids, cassette vectors have been described that serve as ready sources for the required origins of transfer (Citation30,Citation92,Citation93). Depending on how the transfer functions are supplied, biparental, triparental, or fourparental matings can be used for conjugal plasmid transfer. In biparental mating, specialized E. coli strains (Citation42,Citation94) supply the transfer functions in trans via a chromosomally integrated copy of RP4. In triparental mating, the transfer functions used for transfer of the mobilizable plasmid from a separate donor strain to a recipient strain are provided by a helper plasmid (Citation95). Fourparental mating is a variation of triparental mating in which two transferable plasmids are transferred from two separate donor strains into a recipient strain with the aid of a helper plasmid–carrying strain (Citation96).
Promoters
Antibiotic resistance genes are often derived from broad-spectrum resistance elements and, therefore, are generally active in most bacteria. But this is not always the case. Other promoters (e.g., lac, tac, trc, T7 and others; reviewed in References Citation32 and Citation33) were derived for expression in E. coli and may or may not exhibit sufficient activity in other bacterial hosts. For some bacteria, especially those with either very low or very high G + C content, species- or subspecies-specific promoters may have to be developed (Citation48,Citation97,Citation98). This can be done intuitively by choosing either promoters of genes that are naturally expressed at constitutive high levels in virtually all bacteria studied to date, for example, groE (Citation71), acp (acyl carrier protein) (Citation71), ribosomal gene promoters (Citation48), or highly expressed genes identified in microarray experiments. While such exercises are relatively easy for identification of constitutive promoters, identification of promoters useful for regulated gene expression is considerably more complicated. Thankfully, however, there is a considerable body of literature where well-characterized regulated expression systems have been shown to be applicable in a host of bacteria other than E. coli, such as the lac promoter and its derivatives (reviewed in References Citation32 and Citation33), arabinose promoter (Citation36,Citation99), rhamnose promoter (Citation37), organic solvent-responsive promoters, and others (reviewed in References Citation32 and Citation33).
Generation of Chromosomal Mutants
Besides cloning and expression of genes in either the native hosts or heterologous hosts, gene function studies necessitate generation of chromosomal mutants and assessment of their resulting phenotypes, if any. Chromosomal mutants can either be generated randomly or site-specifically. Arguably the most powerful method for generating random pools of chromosomal mutants are transposon-based and such methodologies have been used to generate genome-wide mutant libraries (Citation100–103). Tn5- (Citation100)(Citation103–105) and Himar1-based (Citation97,Citation102)(Citation106–108) elements transpose readily and randomly in most bacteria, but may necessitate some engineering to increase transposition and selection efficiencies. for increased versatility, the transposons may be equipped with replicons (Citation109,Citation110), affinity tags, and reporter genes (Citation101)(Citation110–112) to facilitate downstream experiments, such as rescue of the transposon and localization of its insertion site, antibody detection of tagged gene products, and protein localization or gene expression studies.
Site-specific mutagenesis is achieved via allele or gene replacement, which can be achieved by one of two methods: linear DNA fragment transfer or employment of a suicide plasmid. For linear DNA fragment transfer, a mutagenic DNA fragment is assembled by PCR (Citation48,Citation98). This fragment contains a selection marker flanked by DNA segments that are homologous to chromosomal target sequences. For λ Red recombinase-mediated recombination, the flanking DNA segments can be shorter than 100 bp but for RecA-mediated recombination using the host’s Rec machinery, they must be considerably longer (up to 1 kb). In E. coli (Citation113) and closely related organisms (Citation114,Citation115), the linear PCR fragment is electroporated into a host strain that expresses the phage λ Red recombinase, which is synthesized under the control of an inducible promoter on a low-copy number plasmid. This is followed by selection of the marker expressed from the PCR fragment. The plasmid expressing the Red recombinase is then cured from the host cell resulting in a marked mutant. Because the antibiotic resistance markers are flanked by FRT sites, unmarked mutants only containing a short FRT scar sequence can be derived by Flp recombinase-mediated marker excision (Citation113). A recent study indicates that the λ Red recombinase system may also be adaptable to bacteria other than E. coli, for example, P. aeruginosa (Citation116).
In naturally transformable bacteria, the linear recombinant DNA fragment is added to competent cells, followed by selection of the marker expressed from the PCR fragment. Transformants are the result of recombination into the homologous target sequences on the chromosome catalyzed by the cellular recombination machinery. If the antibiotic marker used for selection is flanked by Cre or Flp recombination sites, unmarked mutants containing either short loxP or FRT scar sequences can be obtained by temporarily providing the respective site-specific recombinases (Citation48,Citation113). Targeted mutagenesis with PCR fragments has most recently been described for Burkolderia pseudomallei (Citation48).
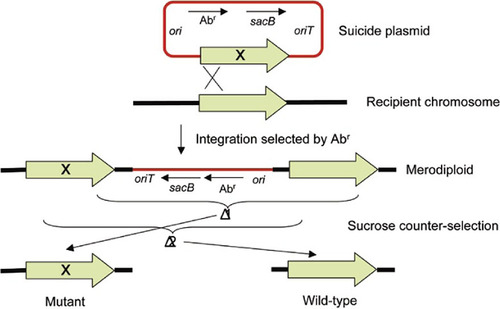
Gene replacement in most bacteria is achieved by cloning the mutated gene of interest into a suicide vector, which is then transferred into the target host, either by electroporation or, more frequently, by conjugation (). In most instances the plasmid backbone also contains a counter-selectable marker to facilitate elimination of unwanted sequences from the frequent merodiploids that result from single-crossover events. Because gene replacement with suicide plasmids usually proceeds via merodiploid formation, mutations in cloned genes don’t necessarily have to be marked with an antibiotic resistance cassette because single-crossover events can be selected by a marker contained on the plasmid backbone. These merodiploids are then resolved in the absence of antibiotic selection leading to a mixed mutant and wild-type cell population that must be distinguished either by phenotype (e.g., growth characteristics) or genotype (e.g., PCR). This method allows generation of unmarked deletion mutants without the scar sequences left behind by site-specific recombinases. It also permits transfer of screenable single nucleotide alleles (e.g., those causing a Ts phenotype) into chromosomal genes. The latter cannot be achieved by fragment mutagenesis because that would necessitate coupling of the mutated allele to a selection marker, which, by definition, would result in two mutations being introduced into the chromosome. A number of different allele replacement vectors containing different counter-selection markers have been described (reviewed in References Citation32 and Citation33).
For allele replacement, the indicated suicide plasmid containing an origin of conjugal transfer (oriT), an antibiotic resistance selection marker (Abr), a narrow-host-range of replication (ori; e.g., pMB1-derived), which is suicidal in most bacteria other than Escherichia coli and its close relatives, a counter-selection marker (here Bacillus subtilis sacB conferring sucrose sensitivity in Gram-negative cells grown in the presence of sucrose), and the mutated gene (the mutation—e.g., single nucleotide change, or in-frame deletion or insertion—is indicated with an X) is transferred into the recipient by conjugation. Selection for Abr leads to merodiploid formation via homologous recombination. Sucrose counter-selection in the absence of antibiotic selective pressure promotes excision of sequences by the indicated deletion events. Deletion 1 causes Rec-mediated excision of undesired plasmid sequences and the wild-type allele, thus resulting in transfer (exchange) of the mutant allele to the chromosome. Deletion 2 also leads to excision of unwanted plasmid sequences but also removes the mutant allele, thus resulting in reversion to wild-type. The two possible outcomes can either be distinguished by a screenable phenotype (e.g., temperature sensitivity) or by genotypic analysis (e.g., PCR). Please note that if the mutated gene is marked with an Abr gene, maintenance of selective pressure with this antibiotic during the counter-selection step will only enable deletion 1 and thus only yield the desired mutant genotype. If this second Abr gene is flanked by recombinase target sites, the marker can be deleted via site-specific marker excision, which will add a couple of extra steps (introduction and curing of a recombinase-encoding plasmid).
Conclusion
The completion of hundreds of genome sequences provides a wealth of information about the virulence and biotechnological and adaptive potential of bacteria. Much of this information has not yet been studied in great detail. However, maximal exploitation of the wealth of information contained in these genomes will necessitate development of genetic tools for many of these less-characterized organisms. The experiences of the past 25 years have taught us that while the focus of genetic tool development has traditionally been driven by studies of model organisms such as E. coli and B. subtilis, steady progress has been made in the development of tools for the manipulation of other bacteria. The challenge of the future will be to steadily improve the arsenal of tools available for genetic studies of some of the fascinating organisms where little but their sequences are known, including yet to be discovered emerging infectious disease pathogens. As evidenced by the reference list of this paper, BioTechniques has played an integral part in the publication of important technological advances in the field over the first 25 years of its existence. Some of these have impressive citation records. For example, a short paper describing small broad-host-range gentamicin resistance cassettes for site-specific insertion and deletion mutagenesis, published by the author in 1993 (Citation49), has been cited no fewer than 246 times as of the time of this review (ISI Web of Knowledge; February 2008).
Competing Interests Statement
The author declares no competing interests.
Acknowledgements
I acknowledge the many undergraduate and graduate students, research associates and postdoctoral fellows that over the years have made valuable contributions to the development of novel genetic tools for diverse bacteria in my laboratory. Supervision of many students would not have been possible without the enduring and expert assistance of Dr. RoxAnn Karkhoff-Schweizer. Financial support to H.P.S. was provided by research grants from the National Institutes of Health and the Cystic Fibrosis Foundation.
Additional information
Funding
References
- Mullis, K.B. and F.A.Faloona. 1987. Specific synthesis of DNA in vitro via a polymerase-catalyzed chain reaction. Methods Enzymol.155:335–350.
- Arnheim, N. and H.Erlich. 1992. Polymerase chain reaction strategy. Annu. Rev. Biochem.61:131–156.
- Scharf, S.J., G.T.Horn, and H.A.Erlich. 1986. Direct cloning and sequence analysis of enzymatically amplified genomic sequences. Science233:1076–1078.
- Horton, R.M., H.D.Hunt, S.N.Ho, J.K.Pullen, and L.R.Pease. 1989. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene77:61–68.
- Ho, S.N., H.D.Hunt, R.M.Horton, J.K.Pullen, and L.R.Pease. 1989. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene77:51–59.
- Fleischmann, R.D., M.D.Adams, O.White, R.A.Clayton, E.F.Kirkness, A.R.Kerlavage, C.J.Bult, J.-F.Tomb, et al.. 1995. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science269:496–512.
- Balbas, P., X.Soberon, E.Merino, M.Zurita, Z.Lomeli, F.Valle, N.Flores, and F.Bolivar. 1986. Plasmid vector pBR322 and its special purpose derivatives—a review. Gene50:3–40.
- Vieira, J. and J.Messing. 1982. The pUC plasmids, an M13mp7-derived system for insertion mutagenesis and sequencing with synthetic universal primers. Gene19:259–268.
- Vieira, J. and J.Messing. 1991. New pUC-derived cloning vectors with different selectable markers and DNA replication origins. Gene100:189–194.
- Prentki, P. 1992. Nucleotide sequence of the classical lacZ deletion ΔM15. Gene122:231–232.
- Alting-Mees, M.A., J.A.Sorge, and J.M.Short. 1992. pBluescript II: multifunctional cloning and mapping vectors. Methods Enzymol.216:483–495.
- Chang, A.C.Y. and S.N.Cohen. 1978. Construction and characterization of amplifiable multicopy DNA cloning vehicles derived from the P15A cryptic miniplasmid. J. Bacteriol.134:1141–1156.
- Martinez, E., B.Bartolome, and F.de la Cruz. 1988. pACYC184-derived cloning vectors containing the multiple cloning site and lacZα reporter gene of pUC8/9 and pUC18/19 plasmids. Gene68:159–162.
- Bartolome, B., Y.Jubete, E.Martinez, and F.de la Cruz. 1991. Construction and properties of a family of pACYC184-derived cloning vectors compatible with pBR322. Gene102:75–78.
- Cohen, S.N., A.C.Chang, H.W.Boyer, and R.B.Helling. 1973. Construction of biologically functional bacterial plasmids in vitro. Proc. Natl. Acad. Sci. USA70:3240–3244.
- Wang, R.F. and S.R.Kushner. 1991. Construction of versatile low-copy-number vectors for cloning, sequencing and gene expression in Escherichia coli. Gene100:195–199.
- Zhou, M.-Y., S.E.Clark, and C.E.Gomez-Sanchez. 1995. Universal cloning method by TA strategy. BioTechniques19:34–35.
- Shuman, S. 1994. Novel approach to molecular cloning and polynucleotide synthesis using vaccinia DNA topoisomerase. J. Biol. Chem.269:32678–32684.
- Katzen, F. 2007. Gateway recombinational cloning: a biological operating system. Expert Opin. Drug Discov.2:571–589.
- Olsen, R.H., G.DeBusscher, and W.R.McCombie. 1982. Development of broad-host-range vectors and gene banks: self-cloning of the Pseudomonas aeruginosa PAO chromosome. J. Bacteriol.150:60–69.
- Pomerantsev, A.P., M.Obuchi, and Y.Ohara. 2001. Nucleotide sequence, structural organization, and functional characterization of the small recombinant plasmid pOM1 that is specific for Francisella tularensis. Plasmid46:86–94.
- Antoine, R. and C.Locht. 1992. Isolation and molecular characterization of a novel broad-host-range plasmid from Bordetella bronchiseptica with sequence similarities to plasmids from gram-positive organisms. Mol. Microbiol.6:1785–1799.
- Pansegrau, W., E.Lanka, P.T.Barth, D.H.Figurski, D.G.Guiney, D.Hass, D.R.Helinski, H.Schwab, et al.. 1994. Complete nucleotide sequence of Birmingham IncP alpha plasmids. Compilation and comparative analysis. J. Mol. Biol.239:623–663.
- Scholz, P., V.Haring, B.Wittmann-Liebold, K.Ashman, M.Bagdasarian, and E.Scherzinger. 1989. Complete nucleotide sequence and gene organization of the broad-host-range plasmid RSF1010. Gene75:271–288.
- Itoh, Y., J.M.Watson, D.Haas, and T.Leisinger. 1984. Genetic and molecular characterization of the Pseudomonas plasmid pVS1. Plasmid11:206–220.
- Kovach, M.E., R.W.Phillips, P.H.Elzer, R.M.RoopII, and K.M.Peterson. 1994. pBBR1MCS: a broad-host-range cloning vector. BioTechniques16:800–802.
- Kovach, M.E., P.H.Elzer, D.S.Hill, G.T.Robertson, M.A.Farris, R.M.Roop, and K.M.Peterson. 1995. Four new derivatives of the broad-host-range cloning vectors pBBR1MCS carrying different antibiotic-resistance cassettes. Gene166:175–176.
- Schweizer, H.P. 1991. Escherichia-Pseudomonas shuttle vectors derived from pUC18/19. Gene97:109–112.
- West, S.E., H.P.Schweizer, C.Dall, A.K.Sample, and L.J.Runyen-Janecky. 1994. Construction of improved Escherichia-Pseudomonas shuttle vectors derived from pUC18/19 and sequence of the region required for their replication in Pseudomonas aeruginosa. Gene148:81–86.
- Schweizer, H.P., T.R.Klassen, and T.Hoang. 1996. Improved methods for gene analysis and expression in Pseudomonas, p. 229–237. In T.Nakazawa, K.Furukawa, D.Haas, and S.Silver (Eds.), Molecular Biology of Pseudomonads. ASM Press, Washington, DC.
- Cronin, C.N. and W.S.McIntire. 1999. pUCP-Nco and pUCP-Nde: Escherichia-Pseudomonas shuttle vectors for recombinant protein expression in Pseudomonas. Anal. Biochem.272:112–115.
- Davison, J. 2002. Genetic tools for pseudomonads, rhizobia, and other Gram-negative bacteria. BioTechniques32:386–401.
- Schweizer, H.P. and V.de Lorenzo. 2004. Molecular tools for genetic analysis of pseudomonads, p. 317–350. In J.L.Ramos(Ed.), Pseudomonas, Vol. 1. Genomics, Life Style and Molecular Architecture. Kluwer Academic/Plenum, , NY.
- Warren, J.W., J.R.Walker, J.R.Roth, and E.Altman. 2000. Construction and characterization of a highly regulable expression vector, pLAC11, and its multipurpose derivatives, pLAC22 and pLAC33. Plasmid44:138–151.
- Guzman, L.-M., D.Belin, M.J.Carson, and J.Beckwith. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol.177:4121–4130.
- Sukchawalit, R., P.Vattanaviboon, R.Sallabhan, and S.Mongkolsuk. 1999. Construction and characterization of regulated L-arabinose-inducible broad host range expression vectors in Xanthomonas. FEMS Microbiol. Lett.181:217–223.
- Cardona, S.T. and M.A.Valvano. 2005. An expression vector containing a rhamnose-inducible promoter provides tightly regulated gene expression in Burkholderia cenocepacia. Plasmid54:219–228.
- Bertani, I., G.Devescovi, and V.Venturi. 1999. Controlled specific expression and purification of 6 × His-tagged proteins in Pseudomonas. FEMS Microbiol. Lett.179:101–106.
- Chebrou, H., Y.Hurtubise, D.Barriault, and M.Sylvestre. 1999. Heterologous expression and characterization of the purified oxygenase component of Rhodococcus globerulus P6 biphenyl dioxygenase and of chimeras derived from it. J. Bacteriol.181:4805–4811.
- Choi, K.-H., L.A.Trunck, A.Kumar, T.Mima, R.R.Karkhoff-Schweizer, and H.P.Schweizer. 2008. Genetic tools for Pseudomonas, p. 65–86. In P.Cornelis(Ed.), Pseudomonas: Genomics and Molecular Biology. Calister Academic Press, Norfolk, UK.
- de Lorenzo, V., M.Herrero, U.Jakubzik, and K.N.Timmis. 1990. Mini-Tn5 transposon derivatives for insertion mutagenesis, promoter probing, and chromosomal insertion of cloned DNA in Gram-negative bacteria. J. Bacteriol.172:6568–6572.
- de Lorenzo, V. and K.N.Timmis. 1994. Analysis and construction of stable phenotypes in Gram-negative bacteria with Tn5 and Tn10-derived transposons. Methods Enzymol.235:386–405.
- McKenzie, G.J. and N.L.Craig. 2006. Fast, easy and efficient: site-specific insertion of transgenes into enterobacterial chromosomes using Tn7 without need for selection of the insertion event. BMC Microbiol.6:39.
- Choi, K.-H., J.B.Gaynor, K.G.White, C.Lopez, C.M.Bosio, R.R.Karkhoff-Schweizer, and H.P.Schweizer. 2005. A Tn7-based broad-range bacterial cloning and expression system. Nat. Methods2:443–448.
- Peters, J.E. and N.L.Craig. 2001. Tn7: smarter than we thought. Nat. Rev. Mol. Cell Biol.2:806–814.
- Choi, K.-H. and H.P.Schweizer. 2006. mini-Tn7 insertion in bacteria with secondary, non-glmS-linked attTn7 sites: example Proteus mirabilis HI4320. Nat. Protoc.1:170–178.
- Choi, K.-H., D.DeShazer, and H.P.Schweizer. 2006. mini-Tn7 insertion in bacteria with multiple glmS-linked attTn7 sites: example Burkholderia mallei ATCC 23344. Nat. Protoc.1:162–169.
- Choi, K.-H., T.Mima, Y.Casart, D.Rholl, I.R.Beacham, and H.P.Schweizer. 2008. Genetic tools for select agent compliant manipulation of Burkholderia pseudomallei. Appl. Environ. Microbiol.74:1064–1075.
- Schweizer, H.P. 1993. Small broad host-range gentamycin resistance cassettes for site-specific insertion and deletion mutagenesis. BioTechniques15:831–834.
- Alexeyev, M.F. 1995. Three kanamycin resistance gene cassettes with different polylinkers. BioTechniques18:52–56.
- Alexeyev, M.F., I.N.Shokolenko, and T.P.Croughan. 1995. Improved antibiotic-resistance gene cassettes and omega elements for Escherichia coli vector construction and in vitro deletion/insertion mutagenesis. Gene160:63–67.
- Deshazer, D. and D.E.Woods. 1996. Broad-host-range cloning and cassette vectors based on the R388 trimethoprim resistance gene. BioTechniques20:762–764.
- Dennis, J.J. and G.J.Zylstra. 1998. Improved antibiotic-resistance cassettes through restriction site elimination using Pfu DNA polymerase PCR. BioTechniques25:772–776.
- Poteete, A.R., C.Rosadini, and C.St Pierre. 2006. Gentamicin and other cassettes for chromosomal gene replacement in Escherichia coli. BioTechniques41:261–264.
- Sadowski, P. 1995. The Flp recombinase of the 2-µm plasmid of Saccharomyces cerevisiae. Prog. Nucleic Acids Res. Mol. Biol.51:53–91.
- Schweizer, H.P. 2003. Applications of the Saccharomyces cerevisiae Flp/FRT system in bacterial genetics. J. Mol. Microbiol. Biotechnol.5:67–77.
- Sternberg, N. and D.Hamilton. 1981. Bacteriophage P1 site-specific recombination. I. Recombination between loxP sites. J. Mol. Biol.150:467–486.
- Sternberg, N., D.Hamilton, and R.Hoess. 1981. Bacteriophage P1 site-specific recombination. II. Recombination between loxP and the bacterial chromosome. J. Mol. Biol.150:487–507.
- Abremski, K., A.Wierzbicki, B.Frommer, and R.H.Hoess. 1986. Bacteriophage P1 Cre-loxP site-specific recombination. Site-specific DNA topoisomerase activity of the Cre recombination protein. J. Biol. Chem.261:391–396.
- Hoang, T.T., R.R.Karkhoff-Schweizer, A.J.Kutchma, and H.P.Schweizer. 1998. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene212:77–86.
- Marx, C.J. and M.E.Lidstrom. 2002. Broad-host-range cre-lox system for antibiotic marker re-cycling in Gram-negative bacteria. BioTechniques33:1062–1067.
- Quenee, L., D.Lamotte, and B.Polack. 2005. Combined sacB-based negative selection and cre-lox antibiotic marker recycling for efficient gene deletion in Pseudomonas aeruginosa. BioTechniques38:63–67.
- Kagle, J. and A.Hay. 2002. Construction of a broad host range cloning vector conferring triclosan resistance. BioTechniques33:490–492.
- Sanchez-Romero, J.M., R.Diaz-Orejas, and V.De Lorenzo. 1998. Resistance to tellurite as a selection marker for genetic manipulations of Pseudomonas strains. Appl. Environ. Microbiol.64:4040–4046.
- Kok, M., M.Rekik, B.Witholt, and S.Harayama. 1994. Conversion of pBR322-based plasmids into broad-host-range vectors by using the Tn3 transposition mechanism. J. Bacteriol.176:6566–6571.
- Vogel, H.J. and D.M.Bonner. 1956. Acetylornithinase of Escherichia coli: partial purification and some properties. J. Biol. Chem.218:97–106.
- Metcalf, W.W., W.Jiang, L.L.Daniels, S.-K.Kim, A.Haldimann, and B.L.Wanner. 1996. Conditionally replicative and conjugative plasmids carrying lacZα for cloning, mutagenesis, and allele replacement in bacteria. Plasmid35:1–13.
- Le Borgne, S., B.Palmeros, F.Valle, F.Bolivar, and G.Gosset. 1998. pBRINT-Ts: a plasmid family with a temperature sensitive replicon, designed for chromosomal integration into the lacZ gene of Escherichia coli. Gene223:213–219.
- Weaver, K.E., K.D.Walz, and M.S.Heine. 1998. Isolation of a derivative of Escherichia coli-Enterococcus faecalis shuttle vector pAM401 temperature sensitive for maintenance in E. faecalis and its use in evaluating the mechanism of pAD1 par-dependent plasmid stabilization. Plasmid40:225–232.
- Phillips, G.J. 1999. New cloning vectors with temperature-sensitive replication. Plasmid41:78–81.
- Maier, T.M., A.Havig, M.Casey, F.E.Nano, D.W.Frank, and T.C.Zahrt. 2004. Construction and characterization of a highly efficient Francisella shuttle plasmid. Appl. Environ. Microbiol.70:7511–7519.
- Gay, P., D.Le Coq, M.Steinmetz, E.Ferrari, and J.A.Hoch. 1983. Cloning structural gene sacB, which codes for exoenzyme levansucrase of Bacillus subtilis: expression of the gene in Escherichia coli. J. Bacteriol.153:1424–1431.
- Ried, J.L. and A.Collmer. 1987. An nptI-sacB-sacR cartridge for constructing directed, unmarked mutations in Gram-negative bacteria. Gene57:239–246.
- Hynes, M.F., J.Quandt, M.P.O’Connell, and A.Puehler. 1989. Direct selection for curing and deletion of Rhizobium plasmids using transposons carrying the Bacillus subtilis sacB gene. Gene78:111–120.
- Blomfield, I.C., V.Vaughn, R.F.Rest, and B.I.Eisenstein. 1991. Allelic exchange in Escherichia coli using the Bacillus subtilis sacB gene and a temperature-sensitive pSC101 replicon. Mol. Microbiol.5:1447–1457.
- Schweizer, H.P. 1992. Allelic exchange in Pseudomonas aeruginosa using novel ColE1-type vectors and a family of cassettes containing a portable oriT and the counter-selectable Bacillus subtilis sacB marker. Mol. Microbiol.6:1195–1204.
- Golovliov, I., A.Sjostedt, A.Mokrievich, and V.Pavlov. 2003. A method for allelic replacement in Francisella tularensis. FEMS Microbiol. Lett.222:273–280.
- Quenee, L., D.Lamotte, and B.Polack. 2005. Combined sacB-based negative selection and cre-lox antibiotic marker recycling for efficient gene deletion in Pseudomonas aeruginosa. BioTechniques38:63–67.
- Stibitz, S. 1994. Use of conditionally counter-selectable suicide vectors for allelic exchange. Methods Enzymol.235:458–465.
- Skrzypek, E., P.L.Haddix, G.V.Plano, and S.C.Straley. 1993. New suicide vector for gene replacement in Yersinia and other gram-negative bacteria. Plasmid29:160–163.
- Kast, P. 1994. pKSS—a second-generation general purpose cloning vector for efficient positive selection of recombinant clones. Gene138:109–114.
- Rahn, A. and C.Whitfield. 2003. Transcriptional organization and regulation of the Escherichia coli K30 group 1 capsule biosynthesis (cps) gene cluster. Mol. Microbiol.47:1045–1060.
- Li, M.Z. and S.J.Elledge. 2005. MAGIC, an in vivo genetic method for the rapid construction of recombinant DNA molecules. Nat. Genet.37:311–319.
- Kristich, C.J., J.R.Chandler, and G.M.Dunny. 2007. Development of a host-genotype-independent counterselectable marker and a high-frequency conjugative delivery system and their use in genetic analysis of Enterococcus faecalis. Plasmid57:131–144.
- Collier, D.N., C.Spence, M.J.Cox, and P.V.Phibbs. 2001. Isolation and phenotypic characterization of Pseudomonas aeruginosa pseudor-evertants containing suppressors of the catabolite repression control-defective crc-10 allele. FEMS Microbiol. Lett.196:87–92.
- Meng, X., R.M.Smith, A.V.Giesecke, J.K.Joung, and S.A.Wolfe. 2006. Counter-selectable marker for bacterial-based interaction trap systems. BioTechniques40:179–184.
- Zhang, X.Z., X.Yan, Z.L.Cui, Q.Hong, and S.P.Li. 2006. mazF, a novel counter-selectable marker for unmarked chromosomal manipulation in Bacillus subtilis. Nucleic Acids Res.34:e71.
- Reyrat, J.M., V.Pelicic, B.Gicquel, and R.Rappuoli. 1998. Counterselectable markers: untapped tools for bacterial genetics and pathogenesis. Infect. Immun.66:4011–4017.
- Nierman, W.C., D.DeShazer, H.S.Kim, H.Tettelin, K.E.Nelson, T.Feldblyum, R.L.Ulrich, C.M.Ronning, et al.. 2004. Structural flexibility in the Burkholderia mallei genome. Proc. Natl. Acad. Sci. USA101:14246–14251.
- Moore, R.A., D.DeShazer, S.Reckseidler, A.Weissman, and D.E.Woods. 1999. Efflux-mediated aminoglycoside and macrolide resistance in Burkholderia pseudomallei. Antimicrob. Agents Chemother.43:465–470.
- Dubnau, D. 1999. DNA uptake in bacteria. Annu. Rev. Microbiol.53:217–244.
- Hengen, P.N. and V.N.Iyer. 1992. DNA cassettes containing the origin of transfer (oriT) of two broad-host-range transfer systems. BioTechniques13:56–58, 60–62.
- Alexeyev, M.F. and I.N.Shokolenko. 1995. RP4 oriT and RP4 and oriT-R6K oriV DNA cassettes for construction of specialized vectors. BioTechniques19:22–26.
- Simon, R., J.Quandt, and W.Klipp. 1989. New derivatives of transposon Tn5 suitable for mobilization of replicons, generation of operon fusions and induction of genes in Gram-negative bacteria. Gene80:161–169.
- Figurski, D.H. and D.R.Helinski. 1979. Replication of an origin-containing derivative of plasmid RK2 dependent on a plasmid function provided in trans. Proc. Natl. Acad. Sci. USA76:1648–1652.
- Choi, K.-H. and H.P.Schweizer. 2006. mini-Tn7 insertion in bacteria with single attTn7 sites: example Pseudomonas aeruginosa. Nat. Protoc.1:153–161.
- Maier, T.M., R.Pechous, M.Casey, T.C.Zahrt, and D.W.Frank. 2006. In Vivo Himar1-based transposon mutagenesis of Francisella tularensis. Appl. Environ. Microbiol.72:1878–1885.
- Liu, J., X.Zogaj, J.R.Barker, and K.E.Klose. 2007. Construction of targeted insertion mutations in Francisella tularensis subsp. novicida. BioTechniques43:487–491.
- Newman, J.R. and C.Fuqua. 1999. Broad-host-range expression vectors that carry the L-arabinose-inducible Escherichia coli araBAD promoter and the araC regulator. Gene227:197–203.
- Jacobs, M.A., A.Alwood, I.Thaipisuttikul, D.H.Spencer, E.Haugen, S.Ernst, O.Will, R.Kaul, et al.. 2003. Comprehensive transposon mutant library of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA100:14339–14344.
- Lewenza, S., R.K.Falsafi, G.Winsor, W.J.Gooderham, J.B.McPhee, F.S.Brinkman, and R.E.Hancock. 2005. Construction of a mini-Tn5-luxCDABE mutant library in Pseudomonas aeruginosa PAO1: a tool for identifying differentially regulated genes. Genome Res.15:583–589.
- Liberati, N.T., J.M.Urbach, S.Miyata, D.G.Lee, E.Drenkard, G.Wu, J.Villanueva, T.Wei, and F.M.Ausubel. 2006. An ordered, nonredundant library of Pseudomonas aeruginosa strain PA14 transposon insertion mutants. Proc. Natl. Acad. Sci. USA103:2833–2838.
- Gallagher, L.A., E.Ramage, M.A.Jacobs, R.Kaul, M.Brittnacher, and C.Manoil. 2007. A comprehensive transposon mutant library of Francisella novicida, a bioweapon surrogate. Proc. Natl. Acad. Sci. USA104:1009–1014.
- Berg, D.E. 1989. Transposon Tn5, p. 185–210. In D.E.Berg, and M.Howe(Ed.), Mobile DNA. American Society for Microbiology, Washington, DC.
- Goryshin, I.Y., J.Jendrisak, L.M.Hoffman, R.Meis, and W.S.Reznikoff. 2000. Insertional transposon mutagenesis by electroporation of released Tn5 transposition complexes. Nat. Biotechnol.18:97–100.
- Lampe, D.J., B.J.Akerley, E.J.Rubin, J.J.Mekalanos, and H.M.Robertson. 1999. Hyperactive transposase mutants of the Himar1 mariner transposon. Proc. Natl. Acad. Sci. USA96:11428–11433.
- Zhang, J.K., M.A.Pritchett, D.J.Lampe, H.M.Robertson, and W.W.Metcalf. 2000. In vivo transposon mutagenesis of the methanogenic archaeon Methanosarcina acetivorans C2A using a modified version of the insect mariner-family transposable element Himar1. Proc. Natl. Acad. Sci. USA97:9665–9670.
- Rubin, E.J., B.J.Akerley, V.N.Novik, D.J.Lampe, R.N.Husson, and J.J.Mekalanos. 1999. In vivo transposition of mariner-based elements in enteric bacteria and mycobacteria. Proc. Natl. Acad. Sci. USA96:1645–1650.
- Dennis, J.J. and G.J.Zylstra. 1998. Plasposons: modular self-cloning minitransposon derivatives for rapid genetic analysis of Gram-negative bacterial genomes. Appl. Environ. Microbiol.64:2710–2715.
- Bolton, A.J. and D.E.Woods. 2000. Self-cloning minitransposon phoA gene-fusion system promotes the rapid genetic analysis of secreted proteins in Gram-negative bacteria. BioTechniques29:470–474.
- Manoil, C. and B.Traxler. 2000. Insertion of inframe sequence tags into proteins using transposons. Methods20:55–61.
- Manoil, C. 2000. Tagging exported proteins using Escherichia coli alkaline phosphatase gene fusions. Methods Enzymol.326:35–47.
- Datsenko, K.A. and B.L.Wanner. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA97:6640–6645.
- Karlinsey, J.E. 2007. Lambda-Red genetic engineering in Salmonella enterica serovar Typhimurium. Methods Enzymol.421:199–209.
- Ranallo, R.T., S.Thakkar, Q.Chen, and M.M.Venkatesan. 2007. Immunogenicity and characterization of WRSF2G11: a second generation live attenuated Shigella flexneri 2a vaccine strain. Vaccine25:2269–2278.
- Lesic, B. and L.G.Rahme. 2008. Use of the lambda Red recombinase system to rapidly generate mutants in Pseudomonas aeruginosa. BMC Mol. Biol.9:20.

