
Abstract
Protein interactions are critical for normal biological processes and molecular pathogenesis. While it is important to study these interactions, there are limited assays that are performed inside the cell, in the native cell environment, where the majority of protein-protein interactions take place. Here we present a method of studying protein interactions intracellularly using one protein of interest fused to a localization-controllable enhanced GFP (EGFP) construct and the other protein of interest fused to the red fluorescent protein, DsRed. Nuclear translocation of the EGFP construct is induced by addition of a ligand, and the difference in nuclear localization between the induced and noninduced states of the DsRed construct provides an indication of the interaction between the two proteins. This assay, the nuclear translocation assay (NTA), is introduced here as broadly applicable for studying protein interactions in the native environment inside cells and is demonstrated using forms of the coiled-coil domain from the breakpoint cluster region (Bcr) protein.
The Nuclear Option
Protein-protein interaction studies offer insight into the molecular mechanisms that govern nearly every normal cellular process as well as aberrant processes that lead to disease. Although the majority of these interactions occur inside cells, only a limited number of intracellular assays are available for detecting protein-protein interactions. Resonance energy transfer methods, such as Förster resonance energy transfer (FRET), are one option, but these depend on sensitive instruments to detect light emitted at specific wavelengths. Yeast and mammalian two-hybrid systems are also options, but these techniques require expression of at least three proteins, including bait, prey, and reporter—as well as any additional control proteins desired. Searching for a simpler approach for qualitative analysis of intracellular protein-protein interactions, A. Dixon and C. Lim from the University of Utah (Salt Lake City, UT) developed a novel nuclear translocation assay. The assay relies on the use of a protein switch containing a nuclear export signal (NES) and a nuclear localization signal (NLS) along with a dexamethasone binding domain. The NES retains the protein switch in the cytoplasm when ligand is absent, but when ligand binds, it induces a conformational change that exposes the NLS and directs the protein into the nucleus. This translocation is dose-dependent and can be reversed by removing the ligand. For the nuclear translocation assay, the authors fused a protein of interest to EFGP and the protein switch, rendering it both detectable by fluorescence microscopy and able to move into the nucleus when bound to the ligand. A potential interacting protein partner was then fused to DsRed. If the two proteins interact in the cytoplasm, both red and green signals would be detected in the nucleus upon addition of the ligand. Measurements of red signal prior to and after ligand addition enables differentiation of induced protein translocation from those proteins that partially localize to the nucleus under normal conditions. The authors validated the assay with wild-type and mutant forms of the coiled-coil domain from the breakpoint cluster region protein (Bcr), which is thought to contribute to oncogenesis during chronic myelogenous leukemia by mediating oligomerization of Bcr and Abelson kinase protein (Abl). These experiments demonstrated that the nuclear translocation assay exhibited the ability to quantify the amount of protein present in each cell and detect variations in binding and specificity of the mutant coiled coil.
See “The nuclear translocation assay for intracellular protein-protein interactions and its application to the Bcr coiled-coil domain” on page 519.
Introduction
Protein-protein interactions are an essential component of almost every cellular process. These interactions regulate signaling events that result in differentiation, proliferation, regulation of gene transcription, repair mechanisms, and inhibition or induction of apoptosis to name only a few (Citation1). Studying these interactions can provide valuable insights into the molecular mechanisms governing these cellular processes. Furthermore, aberrant protein interactions can lead to diseases such as cancer (Citation2), diabetes (Citation3), cystic fibrosis (Citation4), Alzheimer's disease (Citation5), and a number of metabolic disorders (Citation6). Thus, the understanding of protein interactions is also critical in the understanding of disease and the development of therapeutic interventions to treat them. The majority of these interactions between proteins occur inside cells, and intracellular experimental assays for protein-protein interactions are the most relevant. However, currently there are few assays that provide a means to quantify protein-protein interactions in the native cell environment. Resonance energy transfer (RET), including bioluminescence resonance energy transfer (BRET) (Citation7) and Förster (fluorescence) resonance energy transfer (FRET) (Citation8), is one mechanism for detecting intracellular protein interactions but requires a specific combination of the right donor and acceptor, along with the sensitive detection of light emitted at specific wavelengths. Further, the proper orientation of the donor and acceptor dipoles within the Förster distance must be achieved for effective energy transfer. The two-hybrid system (yeast or mammalian) is another assay for intracellular protein interactions (Citation9), but the interaction typically takes place inside the cell nucleus, and this assay requires the transcription/translation of a reporter protein that necessitates the expression of three proteins (or even four if normalizing to a second control protein). CytoTrap is one variation of the two-hybrid assay that allows the interaction to occur in the cytoplasm. The split ubiquitin assay (Citation10) is similar to the two-hybrid system, but uses truncations of ubiquitin in place of transcription factor domains. Here we present a new method for the qualitative analysis of protein-protein interactions that is performed inside cells and is based on an optimized ligand-responsive protein and the simultaneous quantification of fluorescence intensity from enhanced GFP (EGFP) and the red fluorescent protein, DsRed.
The controlled nuclear translocation in this assay occurs through what we have termed a protein switch (PS). The PSs we have developed are chimeric proteins consisting of three components: (i) a nuclear export signal (NES); (ii) a nuclear localization signal (NLS); and (iii) a ligand binding domain (LBD). In the absence of ligand, the NES directs the PS to the cytoplasm. However, when the ligand binds the LBD, it causes a conformational change, exposes the NLS, and the protein is redirected to the nucleus. In our laboratory, various combinations of these three components have been studied to identify the optimal combination that results in the greatest amount of nuclear translocation upon ligand induction (Citation11,Citation12). This nuclear translocation is concentration-dependent and reversible if the ligand is removed (Citation12). One such optimized PS, used herein, consists of the NES from HIV rev, the NLS from MycA8, and the LBD from the rat glucocorticoid receptor containing the C656G mutation, which renders it 10 times more sensitive to the synthetic glucocorticoid dexamethasone (Citation13). This PS provides an easy method for controlling the subcellular shuttling between the cytoplasm and the nucleus.
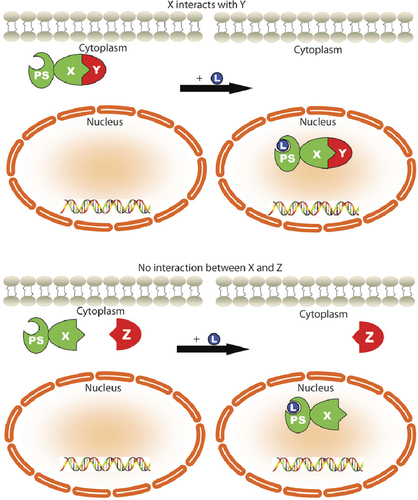
The ability to control the translocation of a protein into the nucleus has led to a biochemical assay for studying protein-protein interactions that we have termed a nuclear translocation assay (NTA). The general concept of the NTA is depicted in . A protein of interest is subcloned and expressed as a fusion protein with the PS and EGFP. As a result of the PS, this fusion protein is responsive to ligand and will translocate into the nucleus upon ligand induction. A second, nonligand-responsive protein tagged with DsRed is co-expressed with the PS construct. If the two proteins interact (, top) the second protein will translocate into the nucleus along with the PS. The percentage of DsRed inside the nucleus is quantified before and after the addition of ligand, and the difference (termed percent nuclear increase, or PNI) is indicative of the interaction between the two proteins. However, if there is little or no interaction between the two proteins (, bottom), the PS alone moves to the nucleus, and there is no change in the subcellular localization of the second protein. Fluorescent protein tags are used as a means of determining the cytoplasmic and nuclear localization of the two proteins through fluorescence microscopy, and thus the fluorescent signals from the two proteins expressed in the same cell need to be distinguishable via excitation/emission filters. Two such fluorescent proteins are EGFP and DsRed, which exhibit excitation/emission of 480 nm/510 nm and 545 nm/620 nm, respectively.
If the two proteins interact, as is the case for proteins X and Y in the top half of the diagram, protein X will cause an increase in the nuclear localization of protein Y upon addition of ligand L, due to the ligand-responsive PS fused to protein X. If two proteins do not interact, as is the case for proteins X and Z in the bottom half of the diagram, the PS translocates the fused protein X to the nucleus upon addition of ligand without altering the localization of protein Y. The translocation of the PS construct is monitored through the fluorescence of EGFP, and the translocation of the second protein is monitored through the fluorescence of DsRed.
To demonstrate the NTA in mammalian cells (1471.1, murine adenocarcinoma; and Cos-7, simian kidney), we performed the assay on two forms of the coiled-coil domain from the breakpoint cluster region (Bcr) protein. This domain forms an antiparallel coiled coil responsible for the oligomerization of Bcr (Citation14). Although relatively little is known about the function of the Bcr protein, a reciprocal chromosomal translocation between chromosomes 9 and 22 generates the Philadelphia chromosome, which expresses Bcr-Abl consisting of the C terminus of Bcr fused to the N terminus of the Abelson kinase protein (Abl) (Citation15). The Philadelphia chromosome and Bcr-Abl are the cause of 95% of chronic myelogenous leukemia (CML) cases (Citation16). Oncogenesis through Bcr-Abl is dependent on the tyrosine kinase activity that is regulated through the oligomerization of Bcr-Abl mediated by the coiled-coil domain (Citation17). Reports have shown that an isolated coiled coil domain (or truncations thereof) can disrupt the oligomerization of Bcr-Abl and reduce the kinase activity and associated oncogenesis (Citation18–21). We have recently altered certain residues (unpublished data) of this domain to prevent the homo-oligomerization (i.e., coiled coil:coiled coil) and improve the binding to Bcr-Abl (i.e., coiled coil:Bcr-Abl). As the wild-type coiled-coil domain (herein designated CC; sequence provided in the Supplementary Materials) has no specificity for Bcr-Abl's coiled coil over another exogenously added coiled coil, the homo-oligomerization may be a limiting factor in its efficacy. Our designed coiled coil (herein designated CCmut; sequence provided in the Supplementary Materials) shows reduced homo-oligomerization (mutant:mutant) along with the potential for enhanced hetero-oligomerization (mutant:Bcr-Abl) to increase the binding to the coiled coil of Bcr-Abl.
The NTA is a unique method for studying protein-protein interactions under intracellular conditions. Few assays offer the ability to study these interactions in the cellular environment where they actually take place. The controlled nuclear translocation upon addition of ligand using the PS provides the ability to differentiate the nuclear translocation from proteins that localize partially in the nucleus or small proteins that can diffuse into the nucleus, by taking the difference in the nuclear percentage before and after ligand induction. Thus, proteins <40 kDa that can diffuse through the nuclear pores or nuclear localized proteins will not result in false positives. However, as this assay revolves around nuclear translocation, transmembrane proteins may not be suitable, and nuclear localized proteins may need a modification (mutating the nuclear localization signal, adding a nuclear export signal, or performing the assay with only the binding domain rather than the full protein) for their study. An additional consideration is that the cytoplasmic and nucleoplasmic environments are not the same, and an interaction that normally occurs in the nucleus may be altered in the cytoplasm where this assay originates. Yet despite these few limitations, the NTA is relatively simple, does not involve reporter plasmids/proteins, and requires little more than the appropriate fusion proteins, dexamethasone, and a fluorescence microscope to perform.
Materials and methods
Construction of plasmids and mutagenesis
The DNA encoding the coiled-coil domain was amplified through PCR from pEGFP-Bcr-Abl using the primers 5′-TGTAACTCGAGTTATGGTGGACCCGGTG-3′ and 5′-ATGCTCTCGAGACCGGTCATAGCTCTTC-3′ and subcloned into pEGFP-PS at the XhoI restriction enzyme site. pEGFP-PS is an EGFP mammalian expression plasmid (pEGFP-C1; Clontech, Mountain View, CA, USA) with the optimized PS subcloned at the C terminus of EGFP (Citation12). Using the resulting pEGFP-PS-CC plasmid as the template, two sequential site-directed mutagenesis reactions were performed using the QuikChange II Site-Directed Mutagenesis kit (Stratagene, La Jolla, CA, USA). The primers used for the first reaction were 5′-GGAGCGCTGCAAGGCCCGCATTCGGCGCGACGAGCAGCGGGACAACCAGGAGCGCTTCCGCATGATCTACCTGGAGACGTTGCTGGCCAAGG-3′ and the reverse complement. For the second reaction, the primers were 5′-GCGACGAGCAGCGGGTGAACCAGGAGCGCTTCC-3′ (and reverse complement) and 5′-CAGGAGCTGGAGCGCGCCAAGGCCCGCATTCG-3′ (and reverse complement). These mutations were designed to change bases encoding five amino acids in the coiled-coil domain to increase the number of ionic interactions in the heterodimer (mutant:wild-type) and introduce charge-charge repulsions in the mutant homodimer (mutant:mutant) (unpublished data). The wild-type and mutant forms of the coiled coil were used as the templates for PCR with the primers 5′-TGTAACTCGAGTTATGGTGGACCCGGTG-3′ and 5′-ATGCTCTCGAGCCGGTCATAGCTCTTC-3′ and subsequently inserted into pDsRed2-N1 (Clontech) at the XhoI site.
Cell lines and transient transfection
1471.1 and Cos-7 cells grow as monolayers in DMEM and RPMI (Gibco, Invitrogen, Carlsbad, CA, USA), respectively, both supplemented with 10% FBS (Hyclone Laboratories, Logan, UT, USA), 1% penicillin-streptomycin (Gibco), 0.1% gentamicin (Hyclone Laboratories), and 1% L-glutamine (Hyclone Laboratories). The cells were maintained in a 5% CO2 incubator at 37°C. 1471.1 cells were grown to ∼50% confluency, and 5 × 106 cells in 100 µL cold plain DMEM were transiently cotransfected via electroporation as previously described (Citation22,Citation23). Transfections were performed using the Electrosquare Porator ECM 830 (BTX, Harvard Apparatus, Holliston, MA, USA) with 2 µg EGFP-PS plasmid and 2 µg DsRed plasmid along with 6 µg pGL3basic (Promega, Madison, WI, USA) carrier DNA for a total of 10 µg DNA. Three pulses of 140 V were applied for 10 ms. Following the electroporation, the cells were plated in complete DMEM in a two-well live cell chamber (Lab-tek II chamber slide system; Nalge NUNC, Rochester, NY, USA) and incubated in a 5% CO2 incubator at 37°C overnight. Cos-7 cells were seeded into a two- or four-well live cell chamber 24 h prior to transfection with Lipofectamine LTX (Invitrogen), so the confluency was ∼90% on the day of the transfection. Transfections were carried out as recommended by the supplier.
Ligand induction and fluorescent microscopy
After 24 h, the cells were rinsed with PBS (Gibco) followed by the addition of 2 mL complete phenol red—free DMEM (Gibco). The PS ligand dexamethasone (Sigma-Aldrich, St. Louis, MO, USA) (Citation12) was then added to one well at a final concentration of 200 nM, and the cells were incubated at 37°C for 1–2 h. H33342 nuclear dye (0.5 µL) was added to the Cos-7 cells and incubated at 37°C for another 15 min. Following the incubation, the transfected cells were viewed and photographed using an inverted fluorescence microscope, Olympus IX701F (Scientific Instrument Company, Sunnyvale, CA, USA) with a high-quality narrow band GFP filter (excitation HQ480/20 nm, emission HQ510/20 nm, beam splitter Q495lp; Chroma Technology Corp., Brattleboro, VT, USA) and a high-quality TRITC filter (excitation HQ545/30 nm, emission HQ620/60 nm, beam splitter Q570lp; Chroma Technology Corp.). Cells were photographed with a 40× or 60× objective using an F-View Monochrome charge-coupled device (CCD) camera. Neutral density filters with 500-ms exposure time were used to minimize photobleaching. The microscope stage was maintained at 37°C with a Nevtek ASI 400 Air Stream Incubator (Nevtek, Williamsville, VA, USA) with temperature control.
Quantification of the PNI
Quantification of the fluorescence intensity of EGFP and DsRed correlates to the amount of the respective exogenous protein. The images of cells were analyzed using analySIS software (Soft Imaging System GmbH, Muenster, Germany), and the amount of protein in the cytoplasm and the nucleus was quantified as previously described (Citation23–26). The following equations were used in the quantification:
where ACI equals the average cytoplasmic intensity, CI equals the cytoplasmic intensity, CA equals the area of cytoplasm, ANI equals the average nuclear intensity, NI equals the nuclear intensity, NA equals the area of nucleus, PN equals the percentage nuclear, PNI equals the percentage nuclear increase, PNL equals the percentage nuclear after ligand induction, and PN0 equals the percentage nuclear without ligand induction.
At least eight cells were analyzed from each group, and each experiment was performed in triplicate. The experimental output, PNI, is a measure of how much the nuclear intensity increases after the addition of ligand, and values reported are the means ± sd. As the PNI of the DsRed construct is related to the binding and PNI of the PS construct, PNI of DsRed was normalized to the PNI of the EGFP (PS) construct, (PNIDsRed/PNIEGFP) × 100%.
Results and discussion
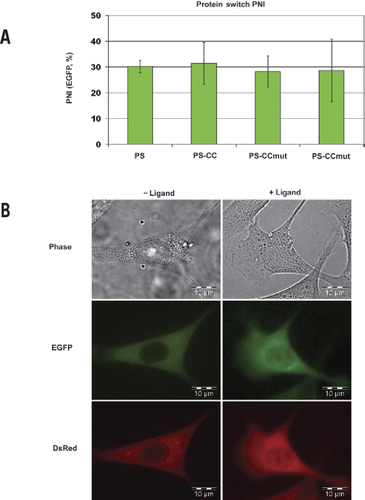
In any assay involving simultaneous expression of two distinct proteins, it is essential to ensure that both proteins are expressed at consistent levels from experiment to experiment and between groups in each experiment. The NTA exhibits the ability to quantify the amount of protein present in each cell being studied, providing a validation of consistent protein expression. For example, the experiments performed in 1471.1 cells all resulted in fluorescence intensities in the range of 13,000 relative fluorescent units (RFU)/area (no statistical difference at the 95% confidence level using one-way ANOVA; data not shown). Further, the EGFP PNI (the ligand-responsive PS) should be relatively consistent and can be monitored to ensure the assay is functioning properly. As seen in , the nuclear fluorescence intensity from all PS constructs (EGFP) assayed in 1471.1 cells increased in the range of 30% after the addition of ligand (no statistical difference at the 95% confidence level using one-way ANOVA). After confirmation that the total fluorescence intensities from both DsRed and EGFP and the PNI of EGFP are consistent, the varying levels of DsRed PNI will be due to the interaction between the two proteins fused to DsRed and EGFP. Example images demonstrating the localization in the absence and presence of ligand in 1471.1 cells are shown in .
(A) PNI of the PS constructs as measured by EGFP fluorescence. Consistent PNI of all PS constructs contributes to validating that the nuclear increase of the DsRed construct is indicative of the interaction. (B) Example images of 1471.1 cells cotransfected with EGFP-PS-CCmut and DsRed-CC. The phase contrast is seen in the top row, the fluorescence from EGFP in the middle row, and fluorescence from DsRed in the bottom row. The left column contains cells without ligand added, and the right column contains cells 2 h after ligand was added. For the PS construct (EGFP), the initial localization is in the cytoplasm, and after addition of ligand, it translocates into the nucleus. The nuclear translocation of the DsRed construct is not as noticeable, but also moves into the nucleus after addition of ligand due to the interaction with the PS construct.
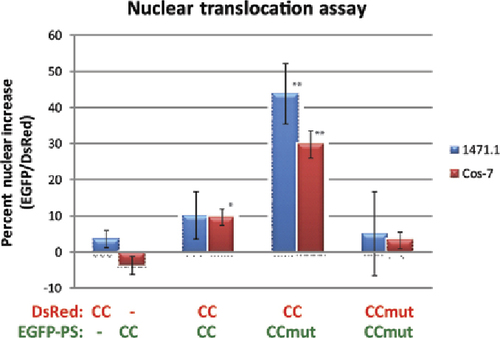
The alteration of subcellular localization of a protein is a novel method of studying protein-protein interactions in the complex cellular milieu. This technology harnesses the control over nuclear translocation provided by the PS and uses the fluorescent proteins EGFP and DsRed to monitor their subcellular localization. As a negative control, EGFPPSCC was contransfected with DsRed (no coiled-coil domain), as well as EGFP-PS (no coiled coil) cotransfected with DsRed-CC, and both resulted in negligible nuclear translocation after the addition of ligand (, first column). A particularly challenging interaction to study is the homo-oligomerization of a protein. When utilizing two fusion proteins to study the interaction, difficulty arises due to the formation of homo-oligomerized fusion proteins that compete with the hetero-oligomerization between the two fusion proteins. In the case of the Bcr coiled-coil domain, in spite of the known dimerization of this domain, the homo-oligomerization of the PS construct seems to be a limiting factor, and minimal nuclear translocation was seen (, second column). However, when we used a mutant form of the coiled coil that exhibits reduced homodimerization, the PS construct was more available for interaction with the DsRed construct, and a large nuclear increase was observed (, third column). The prevention of the homo-oligomerization by the mutant coiled coil is also indicated by the absence of nuclear increase when the mutant coiled coil is fused to both the PS and DsRed (, fourth column). The NTA provided a novel way of demonstrating the enhanced binding and specificity of the mutant coiled coil as seen in . This data has been corroborated with a mammalian two-hybrid assay (data not shown).
The mutant coiled coil is designed to prevent homo-oligomerization (CCmut:CCmut) and to improve binding to the wild-type coiled coil. This results in EGFP-PSCCmut being more available to oligomerize with DsRed-CC and the greatest nuclear translocation with this combination (CCmut:CC, third column). PS, protein switch; CC, coiled coil; CCmut, mutant coiled coil. *P < 0.01, **P < 0.001 compared with control (first column); each experiment was performed in triplicate with at least eight cells analyzed per experiment. Statistical significance was determined using one-way ANOVA with Tukey's post-test.
One of the cell lines we chose to use for this assay, 1471.1, is an adherent cell line with easily detectable nuclei ideal for imaging fluorescent proteins via fluorescence microscopy. These characteristics make this cell line a suitable candidate for the NTA. Further, the cell nucleus is very large (), and the nuclear membrane is easily detectable with a 40× objective, eliminating the need for nuclear stains and reducing the complexity of the assay. We have routinely stained the nuclei of these cells with a Hoechst dye (H33342, similar to DAPI) and confirmed the location of the cell nuclei (data not shown). To further validate the assay, we studied the same interactions in Cos-7 cells, and very similar results were obtained, as seen in .
The PS designed in our laboratory originated with an idea for altering the subcellular localization of aberrant functioning proteins as a therapeutic intervention (Citation23). This optimized control over nuclear translocation now serves as an assay to study protein interactions in the intracellular matrix and can be applied to almost any protein of interest. Although in some regards, the NTA is similar to the membrane recruitment assay (Citation27) or the translocation biosensor (Citation28), the ability to control the translocation by the presence or absence of a ligand is advantageous. Here we have demonstrated the NTA in murine and simian cells, but as the PS is translocated into the nucleus via importin α/β, common among most organisms, other cells (including human) could be used to perform this assay. After routine generation of fusion proteins consisting of the proteins of interest fused to EGFP-PS and DsRed, the NTA is simple and requires only a fluorescence microscope, software to quantify fluorescence intensities (such as Image J freeware; http://rsbweb.nih.gov/ij), and the readily available ligand dexamethasone. This assay now adds to the limited repertoire of experimental techniques for studying protein interactions inside of cells.
Competing interests
The authors declare no competing interests.
Acknowledgments
Funding was provided by the National Institutes of Health (NIH; grant no. R01 CA129528). We acknowledge the use of DNA/Peptide Core supported by the National Cancer Institute (NCI) Cancer Center Support Grant no. P30 CA042014, awarded to Huntsman Cancer Institute. We would like to thank Thomas Cheatham, Scott Pendley, Benjamin Bruno, Jonathan Constance, Rian Davis, Mohanad Mossalam, and David Woessner for scientific input. This paper is subject to the NIH Public Access Policy.
Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at: www.tandfonline.com/doi/suppl/10.2144/000113450
Additional information
Funding
References
- Stein, A., R.A.Pache, P.Bernado, M.Pons, and P.Aloy. 2009. Dynamic interactions of proteins in complex networks: a more structured view. FEBS J.276:5390–5405.
- Davis, J.R., M.Kakar, and C.S.Lim. 2007. Controlling protein compartmentalization to overcome disease. Pharm. Res.24:17–27.
- Tsukaguchi, H., H.Matsubara, S.Taketani, Y.Mori, T.Seido, and M.Inada. 1995. Binding-, intracellular transport-, and biosynthesis-defective mutants of vasopressin type 2 receptor in patients with X-linked nephrogenic diabetes insipidus. J. Clin. Invest.96:2043–2050.
- Buchanan, P.J., R.K.Ernst, J.S.Elborn, and B.Schock. 2009. Role of CFTR, Pseudomonas aeruginosa and Toll-like receptors in cystic fibrosis lung inflammation. Biochem. Soc. Trans.37:863–867.
- Zhong, N. and K.H.Weisgraber. 2009. Understanding the basis for the association of apoE4 with Alzheimer's disease: opening the door for therapeutic approaches. Curr. Alzheimer Res.6:415–418.
- Gloerich, J., R.A.Wevers, J.A.Smeitink, B.G.van Engelen, and L.P.van den Heuvel. 2007. Proteomics approaches to study genetic and metabolic disorders. J. Proteome Res.6:506–512.
- Pfleger, K.D. and K.A.Eidne. 2006. Illuminating insights into protein-protein interactions using bioluminescence resonance energy transfer (BRET). Nat. Methods3:165–174.
- Miyawaki, A., J.Llopis, R.Heim, J.M.McCaffery, J.A.Adams, M.Ikura, and R.Y.Tsien. 1997. Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature388:882–887.
- Suter, B., S.Kittanakom, and I.Stagljar. 2008. Two-hybrid technologies in proteomics research. Curr. Opin. Biotechnol.19:316–323.
- Johnsson, N. and A.Varshavsky. 1994. Split ubiquitin as a sensor of protein interactions in vivo. Proc. Natl. Acad. Sci. USA91:10340–10344.
- Kakar, M., A.B.Cadwallader, J.R.Davis, and C.S.Lim. 2007. Signal sequences for targeting of gene therapy products to subcellular compartments: the role of CRM1 in nucleocytoplasmic shuttling of the protein switch. Pharm. Res.24:2146–2155.
- Kakar, M., J.R.Davis, S.E.Kern, and C.S.Lim. 2007. Optimizing the protein switch: altering nuclear import and export signals, and ligand binding domain. J. Control. Release120:220–232.
- Htun, H., J.Barsony, I.Renyi, D.L.Gould, and G.L.Hager. 1996. Visualization of glucocorticoid receptor translocation and intranuclear organization in living cells with a green fluorescent protein chimera. Proc. Natl. Acad. Sci. USA93:4845–4850.
- Taylor, C.M. and A.E.Keating. 2005. Orientation and oligomerization specificity of the Bcr coiled-coil oligomerization domain. Biochemistry44:16246–16256.
- Rowley, J.D. 1973. Letter: a new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature243:290–293.
- Sandberg, A.A., R.M.Gemmill, B.K.Hecht, and F.Hecht. 1986. The Philadelphia chromosome: a model of cancer and molecular cytogenetics. Cancer Genet. Cytogenet.21:129–146.
- Zhang, X., R.Subrahmanyam, R.Wong, A.W.Gross, and R.Ren. 2001. The NH(2)-terminal coiled-coil domain and tyrosine 177 play important roles in induction of a myeloproliferative disease in mice by Bcr-Abl. Mol. Cell. Biol.21:840–853.
- Guo, X.Y., J.M.Cuillerot, T.Wang, Y.Wu, R.Arlinghaus, D.Claxton, C.Bachier, J.Greenberger, et al.. 1998. Peptide containing the BCR oligomerization domain (AA 1–160) reverses the transformed phenotype of p210bcr-abl positive 32D myeloid leukemia cells. Oncogene17:825–833.
- Beissert, T., A.Hundertmark, V.Kaburova, L.Travaglini, A.A.Mian, C.Nervi, and M.Ruthardt. 2008. Targeting of the N-terminal coiled coil oligomerization interface by a helix-2 peptide inhibits unmutated and imatinib-resistant BCR/ABL. Int. J. Cancer122:2744–2752.
- Beissert, T., E.Puccetti, A.Bianchini, S.Guller, S.Boehrer, D.Hoelzer, O.G.Ottmann, C.Nervi, and M.Ruthardt. 2003. Targeting of the N-terminal coiled coil oligomerization interface of BCR interferes with the transformation potential of BCR-ABL and increases sensitivity to STI571. Blood102:2985–2993.
- Huang, S.F., D.B.Liu, J.M.Zeng, Q.Xiao, M.Luo, W.P.Zhang, K.Tao, J.P.Wen, et al.. 2009. Cloning, expression, purification and functional characterization of the oligomerization domain of Bcr-Abl oncoprotein fused to the cytoplasmic transduction peptide. Protein Expr. Purif.64:167–178.
- Kanwal, C., H.Li, and C.S.Lim. 2002. Model system to study classical nuclear export signals. AAPS PharmSci4:E18.
- Kanwal, C., S.Mu, S.E.Kern, and C.S.Lim. 2004. Bidirectional on/off switch for controlled targeting of proteins to subcellular compartments. J. Control. Release98:379–393.
- Li, H., M.L.Fidler, and C.S.Lim. 2005. Effect of initial subcellular localization of progesterone receptor on import kinetics and transcriptional activity. Mol. Pharm.2:509–518.
- Li, H., G.Yan, S.E.Kern, and C.S.Lim. 2003. Correlation among agonist dose, rate of import, and transcriptional activity of liganded progesterone receptor B isoform in living cells. Pharm. Res.20:1574–1580.
- Lim, C.S., C.T.Baumann, H.Htun, W.Xian, M.Irie, C.L.Smith, and G.L.Hager. 1999. Differential localization and activity of the A- and B-forms of the human progesterone receptor using green fluorescent protein chimeras. Mol. Endocrinol.13:366–375.
- Grefen, C., K.Stadele, K.Ruzicka, P.Obrdlik, K.Harter, and J.Horak. 2008. Subcellular localization and in vivo interactions of the Arabidopsis thaliana ethylene receptor family members. Mol. Plant.1:308–320.
- Knauer, S.K. and R.H.Stauber. 2005. Development of an autofluorescent translocation biosensor system to investigate proteinprotein interactions in living cells. Anal. Chem.77:4815–4820.

