The Missing Link
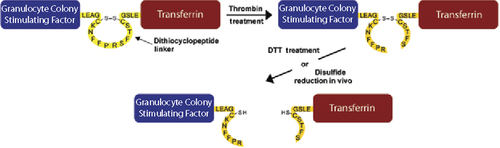
Certain recombinant fusion proteins require short peptide linkers between protein domains to provide flexibility or increase stability during synthesis. For some fusion proteins, however, the inability to cleave these stable peptide linkers in order to separate and release the protein domains can impede their in vivo function. One alternative is to utilize a reversible disulfide linkage between the two protein domains, either through cysteine bonding or bifunctional linkers, but this requires chemical conjugation of the domains and often results in heterogeneous products unsuitable for therapeutic use. In this issue, Chen et al. at the University of Southern California (Los Angeles, CA) describe a novel in vivo cleavable disulfide peptide linker for fusion proteins that overcomes these issues. Their dithiocyclopeptide linker is based on the structure of somatostatin, where two cysteine residues form an intramolecular disulfide bond, resulting in a cyclopeptide. The peptide linker also was modified to contain a thrombin-sensitive sequence between the cysteine residues. While in vitro thrombin treatment of a fusion protein containing this linker results in cleavage of the dithiocyclopeptide linker, the protein domains remain attached through the disulfide bond. After administration into whole animals, in vivo reduction of the disulfide bond separates the two protein domains, allowing them to function independently. The authors demonstrated their method by joining granulocyte colony stimulating factor (G-CSF) and transferrin (Tf) protein domains using the dithiocyclopeptide linker to generate a fusion protein (G-C-T). In vitro, free G-CSF was only released from G-C-T treated with both thrombin and dithiothreitol and not from untreated G-C-T, and the former was twice as biologically active as the latter in a cell proliferation assay. After intravenously injecting mice with thrombin-treated but still—disulfide-intact G-C-T fusion protein, free G-CSF was detected in the blood after only 5 min; peak amounts were measured after 15 min, whereas none was detected in mice injected with untreated G-C-T.
See “Design of an in vivo cleavable disulfide linker in recombinant fusion proteins”.
The Nuclear Option
Protein-protein interaction studies offer insight into the molecular mechanisms that govern nearly every normal cellular process as well as aberrant processes that lead to disease. Although the majority of these interactions occur inside cells, only a limited number of intracellular assays are available for detecting protein-protein interactions. Resonance energy transfer methods, such as Förster resonance energy transfer (FRET), are one option, but these depend on sensitive instruments to detect light emitted at specific wavelengths. Yeast and mammalian two-hybrid systems are also options, but these techniques require expression of at least three proteins, including bait, prey, and reporter—as well as any additional control proteins desired. Searching for a simpler approach for qualitative analysis of intracellular protein-protein interactions, A. Dixon and C. Lim from the University of Utah (Salt Lake City, UT) developed a novel nuclear translocation assay. The assay relies on the use of a protein switch containing a nuclear export signal (NES) and a nuclear localization signal (NLS) along with a dexamethasone binding domain. The NES retains the protein switch in the cytoplasm when ligand is absent, but when ligand binds, it induces a conformational change that exposes the NLS and directs the protein into the nucleus. This translocation is dose-dependent and can be reversed by removing the ligand. For the nuclear translocation assay, the authors fused a protein of interest to EFGP and the protein switch, rendering it both detectable by fluorescence microscopy and able to move into the nucleus when bound to the ligand. A potential interacting protein partner was then fused to DsRed. If the two proteins interact in the cytoplasm, both red and green signals would be detected in the nucleus upon addition of the ligand. Measurements of red signal prior to and after ligand addition enables differentiation of induced protein translocation from those proteins that partially localize to the nucleus under normal conditions. The authors validated the assay with wild-type and mutant forms of the coiled-coil domain from the breakpoint cluster region protein (Bcr), which is thought to contribute to oncogenesis during chronic myelogenous leukemia by mediating oligomerization of Bcr and Abelson kinase protein (Abl). These experiments demonstrated that the nuclear translocation assay exhibited the ability to quantify the amount of protein present in each cell and detect variations in binding and specificity of the mutant coiled coil.
FISH and Chips
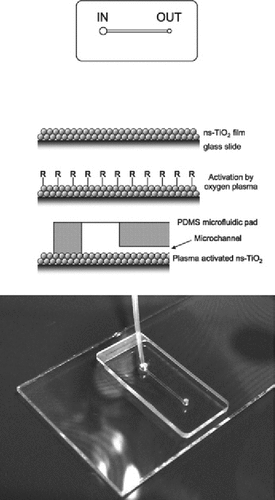
Fluorescence in situ hybridization (FISH) is frequently used in basic research and clinical settings to resolve complex chromosomal rearrangements. While the technique is sensitive and effective, the probes needed for FISH can be costly and samples with reduced numbers of cells may not yield useful results. Theengineering of polymeric microchannels within microfluidic devices offers possible solutions to these problems since smaller volumes of reagents are required and cell samples can be concentrated, but their implementation for FISH experiments has been limited due to the shearing forces generated by solutions moving through the channels. Immobilization of cells within the channels is therefore necessary to ensure that meaningful FISH results can be obtained from cell samples in microfluidic devices. A. Zanardi et al. at Tethis S.R.L. (Milan, Italy) recently found that cluster-assembled nanostructured TiO2 (ns-TiO2) coatings are able to rapidly immobilize living and fixed hematopoietic cells and hold them stably, even in the presence of prolonged shear stresses. Using this coating, the authors have engineered a microfluidic device capable of immobilizing cell samples for miniature FISH. The goals of their design were to decrease the cost of FISH by reducing the required volume of reagents and fluorescent probe, increase the throughput of the experiments by allowing parallel processing of samples and automated microscopic evaluation of the results, and enable FISH with cell-sparse samples by concentrating the cells within a confined space. The device, which successfully fulfilled each of the authors' goals, consists of a single straight microchannel in a polymeric microfluidic pad, which adheres to a standard glass slide coated with ns-TiO2. The device was validated in a series of FISH experiments conducted on cultured hematopoietic tumor cells, bone marrow, and peripheral blood samples and run in parallel with a standard FISH protocol. Using miniaturized FISH, the authors could detect hematological malignancies such as acute lymphoblastic leukemia, chronic lymphocytic leukemia, acute myeloid leukemia, as well as a case of sex chromosome chimerism following bone marrow transplant, all with similar accuracy and reproducibility to standard FISH.
See “Miniaturized FISH for screening of onco-hematological malignancies”.
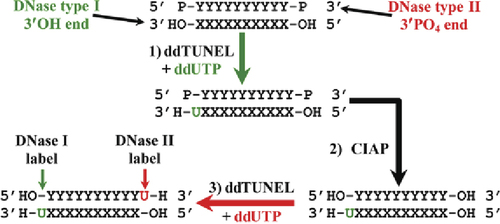
Digging a New TUNEL
The terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay is a standard method for detecting DNA fragmentation associated with apoptosis. Labeled dUTP is added by terminal deoxynucleotidyl transferase to the 3′OH ends of DNA fragments that are generated by type I DNases during apoptosis. However, this assay is inherently nonquantitative since the added dUTP itself has a 3′OH end, resulting in the formation of deoxyuridine polymers of indeterminate length at each primary 3′OH site. In this issue, Baskin et al. from the Methodist Hospital (Houston, T X) present a modified TUNEL assay they call ddTUNEL, which uses ddUTP instead of dUTP to stoichiometrically label each DNA3′OH end with a sing le dideoxyuridine base that cannot be further extended, thereby permitting quantitation of the number of 3′OH ends by fluorescence microscopy. This capping of the DNase type I—induced 3′OH ends has the added benefit of enabling additional rounds of ddTUNEL to examine 3′OH ends subsequently generated from other forms of DNA damage in the same sample. For instance, the 3′ phosphate ends of DNA fragments generated by type II DNases associated with caspase independent apoptosis can be detected by treatment of the sample from a first round of ddTUNEL with calf intestinal alkaline phosphatase (CIAP). This creates additional 3′OH ends that can be differentially labeled in a second round of ddTUNEL. Modified bases and apurinic/apyrimidinic (AP) sites in DNA can also be independently assayed by using the bacterially derived enzyme formamidopyrimidine-DNA glycosylase, which excises many modified bases to form an AP site that is then hydrolyzed to a nick containing a 3′ phosphate end that can then be converted to a 3′OH end with CIAP. These new ddTUNEL assays were validated in a study of mammary involution, which involves large-scale apoptosis, and by an examination of DNA damage in U87 cells upon exposure to various DNA-modifying agents.