
FlAsH Forward
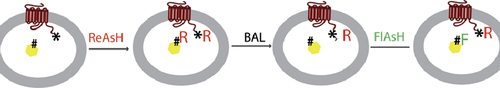
Nijsje DormanPatrick C.H. LoKristie NyboSmall fluorescent labels such as the fluorescein arsenical hairpin binder (FlAsH) make popular alternatives to bulky fluorescent proteins. FlAsH, along with its color variant ReAsH (red arsenical hairpin binder) interacts with a tetra-cysteine motif whose small size minimizes disruption to protein function. In addition (because it doesn't require a flexible linker), the fluorophore is locked into a more easily definable position, a feature that is appealing for Förster resonance energy transfer (FRET) experiments. In previous FRET studies, FlAsH has been used in combination with cyan fluorescent protein (CFP), so the potential limitations associated with fluorescent proteins remain a concern. A more effective system would be to use FlAsH and ReAsH as FRET partners. Although a sequential labeling strategy for these fluorophores has been described in purified proteins, the method didn't address selective labeling of protein sites in intact cells. In Bioconjugate Chemistry, Zürn et al. fill this gap by showing selective labeling of the membrane-bound parathyroid hormone (PTH) receptor and its cytosolic binding partner β-arrestin2. The strategy rests on placing a 12—amino acid high-affinity FlAsH/ReAsH binding motif in one protein, and a lower-affinity binding motif (of 6 amino acids) in the other. After incubation with ReAsH, the cells are washed with dimercaptopropanol, a competitive inhibitor of FlAsH/ReAsH binding. The dimercaptopropanol concentration is set so that the fluorophore's binding to the lower-affinity sequence is nearly fully outcompeted, while its interaction with the high-affinity motif is essentially unaffected. When the cells are subsequently incubated with FlAsH, the lower-affinity motifs become labeled, but the high-affinity motifs remain bound to ReAsH. This simple approach delivers selective labeling with minimal background, the proteins can be tracked by confocal microscopy, and the fluorescent tags do not interfere with normal behavior of the proteins in response to ligand binding to the PTH receptor. Measurements of the emission ratios of FlAsH and ReAsH showed that FRET occurred when colocalization was triggered. The system worked equally well when the binding motifs were swapped between the two proteins, suggesting the domains perform equivalently in different structural contexts. Ongoing development of brighter biarsenical fluorophore tags of various colors should open new opportunities for cell labeling and tracking.
Reprinted with permission © 2010 ACS.
Skip to the End
Nijsje DormanPatrick C.H. LoKristie NyboThe importance of the 3′ untranslated region (UTR) to post-transcriptional regulation of eukaryotic gene expression is increasingly appreciated but hard to study in isolation. A central difficulty is that stimuli that act on UTRs often also affect promoter activity. For instance, AU-rich mRNA destabilizing elements (AREs) respond to cytokines such as tumor necrosis factor α (TNF-α) and interleukin 1-α(IL-1α), both of which induce the cytomegalovirus (CMV) promoter. In an article in RNA, Hitti et al. introduce a new reporter that permits selective examination of the regulatory effect of the 3′ end of the transcript. The authors designed a green fluorescent protein (GFP) reporter construct where expression is driven by an optimized promoter from the ribosomal protein S30 (RPS30), which shows that this modified promoter is impervious to various stress inducers known to act through ARE-mediated pathways. When AREs from IL-8 or TNF-α were placed within the reporter, fluorescence was significantly more reduced (reflecting ARE-mediated transcript destabilization) than in a CMV-driven construct, where the effect was likely obscured by promoter effects. In further studies, the authors explain how comparing RNA level (by quantitative reverse transcription PCR) and protein amount (by GFP fluorescence) helps coax apart transcript stability and translational blockage. The system is also suitable for studying regulation by RNA-binding proteins, and gives more meaningful results than traditional reporter vectors. For example, cotransfection with a plasmid expressing the ARE-binding protein HuR increases signal from a CMV-driven reporter even in the absence of an ARE. In contrast, the modulation of the RPS30-based reporter is ARE-dependent. A helpful feature of the reporter is that transcriptional response elements can easily be appended, permitting study of the combined effects of transcriptional and post-transcriptional modulation. The authors also describe a Tet-Off variant of the modified RPS30 promoter for mRNA half-life determinations without the need for agents such as actinomycin D, which can have nonspecific toxicity. These features make the new reporter a flexible tool for analyzing mRNA regulation, all in the context of a robust promoter and modular structure that simplifies the study of functional elements, particularly in the 3′ end of the transcript.
Creating Photosensitive Fluorescent Proteins: It's a SNAP
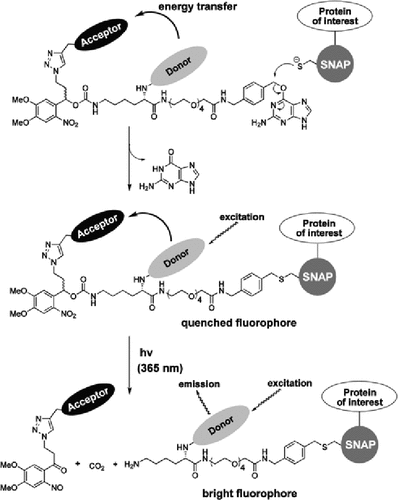
Nijsje DormanPatrick C.H. LoKristie NyboPhotoactivatable (PA) and photoconvertible (PC) fluorescent proteins are invaluable tools in cell biology research, but their wider usage has been hampered by a limited color palette. An alternative to creating new photosensitive proteins is to specifically label proteins with photosensitive fluorophores [e.g., reacting SNAP-tag fusion proteins with derivatives of the SNAP-tag substrate O6-benzylguanine (BG) attached to photosensitive fluorophores]. One versatile approach to creating different PA or PC fluorophores is to attach a donor fluorophore to a quencher or an acceptor fluorophore through a photocleavable linker. A recent paper in Chemical Biology by Maurel et al. combined these two approaches in a flexible method to generate a broad range of PA and PC fluorophores containing BG moieties for attachment to SNAP-tagged fusion proteins. As examples, they created two PA probes (Q-Fl and Q-Cy3) by connecting the quencher QSY7 through a linker to the fluorophore—fluorescein or Cy3, respectively—which is joined to a BG moiety. Photocleavage of the linker in these compound fluorophores releases the quencher, leaving an unquenched fluorescein or Cy3 molecule attached to the SNAP tag. To generate a probe that photoconverts from Cy5 to Cy3 emission, Cy5 was connected through the linker to Cy3 joined to BG. Excitation of Cy3 in this compound fluorophore (Cy5-Cy3) results in Cy5 emission through Förster resonance energy transfer between Cy3 and Cy5, while subsequent photocleavage of the linker releases the Cy5, resulting in Cy3 emission. Since these fluorophores are impermeable to living cells and therefore label only SNAP-tagged cell surface proteins, the approach is useful in studying the dynamics of lateral mobility of surface proteins in the plasma membrane following photoactivation or photoconversion. Intracellular protein labeling could also be achieved by cell loading of the substrates using glass beads, with efficient photoactivation of labeled, SNAP-tagged nuclear and cytoplasmic proteins being observed.
Reprinted with permission © 2010 ACS.
QUBIC
Nijsje DormanPatrick C.H. LoKristie NyboLarge-scale genetic screens have dramatically increased the rate at which genome-wide phenotypic studies can be performed. But techniques to map the protein changes leading to certain observed phenotypes have lagged behind. While interaction mapping of protein complexes can be performed using a combination of affinity purification and mass spectrometry (MS), true interacting partners can be difficult to distinguish from background proteins bound nonspecifically to the antibodies or beads. This results in a high incidence of false-positive interactions, often necessitating more stringent purification methods that may result in the loss of weak or transient protein interactions. The manner in which a protein is expressed can also compromise interaction data, since tagged bait proteins are commonly controlled by plasmid promoters rather than endogenous promoters. In the Journal of Cell Biology, Hubner et al. describe a new platform incorporating quantitative interaction proteomics, bacterial artificial chromosome (BAC) recombineering to facilitate precise manipulation of BAC transgenes for expression of bait proteins in their native context, and GFP affinity tagging to enable live cell imaging for improved large-scale interaction studies in mammalian cells. The technique, which the authors call quantitative BAC-GFP interactomics (QUBIC), is performed using either stable isotope labeling with amino acids in cell culture (SILAC) or label-free approaches. GFP-tagged and wild-type cells are cultured (with differing forms of lysine for the SILAC approach), then separately pulled-down by magnetic beads coupled to anti-GFP antibodies and eluted by in-column digestion. For the label-free approach, eluates can be directly analyzed by MS. And for the SILAC approach, the eluates are merged, identified by high-resolution MS, and quantified by comparison of the relative intensities of the light and heavy forms of each peptide. Using QUBIC, the authors were able to overcome the limitations of current protein interaction screens since the new platform allows endogenous expression and regulation of bait proteins to detect transient or weak interactions, as well as compatibility with live cell imaging methods.

