
Abstract
Current methods for targeted massively parallel sequencing (MPS) have several drawbacks, including limited design flexibility, expense, and protocol complexity, which restrict their application to settings involving modest target size and requiring low cost and high throughput. To address this, we have developed Hi-Plex, a PCR-MPS strategy intended for high-throughput screening of multiple genomic target regions that integrates simple, automated primer design software to control product size. Featuring permissive thermocycling conditions and clamp bias reduction, our protocol is simple, cost- and time-effective, uses readily available reagents, does not require expensive instrumentation, and requires minimal optimization. In a 60-plex assay targeting the breast cancer predisposition genes PALB2 and XRCC2, we applied Hi-Plex to 100 ng LCL-derived DNA, and 100 ng and 25 ng FFPE tumor-derived DNA. Altogether, at least 86.94% of the human genome-mapped reads were on target, and 100% of targeted amplicons were represented within 25-fold of the mean. Using 25 ng FFPE-derived DNA, 95.14% of mapped reads were on-target and relative representation ranged from 10.1-fold lower to 5.8-fold higher than the mean. These results were obtained using only the initial automatically-designed primers present in equal concentration. Hi-Plex represents a powerful new approach for screening panels of genomic target regions.
A trip to the library
Massively parallel sequencing (MPS)—i.e., next generation sequencing—has revolutionized molecular biology, catalyzing the explosive growth of genomics and transcriptomics. Construction of a library from target DNA remains an essential step in most MPS protocols. In these cases, proper library preparation and quality control are critical to obtain the best sequence quality as well as to generate the maximum amount of sequence data. Optimizing library construct requires two critical steps: first, the amount of DNA library molecules loaded must be precisely calibrated, as either too little or too much can compromise sequencing data quality or result in failed sequencing runs; and second, the library molecules need to be in an appropriate size range to maximize sequencing throughput. In this month's issue, two papers from different groups describe new methods to optimize MPS library preparation. In the first article, D.J. Park and colleagues at the University of Melbourne (Melbourne, Australia) present a new methodology called Hi-Plex PCR that simplifies targeted MPS resequencing. Several current targeted PCR-based MPS resequencing methods suffer from the need for the labor-intensive normalization of PCR products prior to sequencing in order to obtain libraries of desired size. In contrast, Hi-Plex provides a customizable approach using simple automated primer design software to control the sizes of the PCR amplicons generated in a single PCR reaction from all the targeted regions, which are then size-selected by gel electrophoresis. This flexibility allows Hi-Plex targeted resequencing to be carried out at much less expense and labor than other such techniques. In the second article, J. Bielas and colleagues at the University of Washington (Seattle, WA) describe an accurate and precise method of quality control for MPS libraries using simultaneous quantification and size determination of amplifiable library molecules by droplet digital PCR (ddPCR). Current protocols for MPS library quality control utilize qPCR for quantification, followed by gel or capillary electrophoresis for size determination. But the qPCR and gel selection steps have various drawbacks, such as amplification biases and the need for standard curves. Bielas' group cleverly demonstrate that ddPCR, which had already been validated for absolute DNA quantification, can also be used simultaneously to calculate the size of target DNA due to a linear correlation the authors discovered between the fluorescence amplitude of a ddPCR droplet and the size of the amplicon within it. Their QuantiSize method is not only more accurate than qPCR for quantification, but also conveniently combines the steps for size and concentration determination into a single assay.
See “A high-plex PCR approach for massively parallel sequencing” on page 69.
Hi-Plex is a streamlined, highly-multiplexed PCR approach for targeted massively parallel sequencing. Our method integrates simple, automated primer design software and simple protocols, requires minimal optimization, does not rely on expensive instrumentation, and uses low-cost, readily available reagents to perform cost-effective and rapid sequencing.
The advent of massively parallel sequencing (MPS) has heralded a new era for genetic testing (reviewed in (Citation1, Citation2). There are a number of potential applications for MPS, including its use on genomic regions selectively captured from a DNA sample before sequencing (targeted MPS). Whole-genome sequencing involves considerably higher costs and lower throughput. Targeted MPS is an attractive approach for the screening of large panels of genes or genomic regions, both in research and diagnostic settings. In candidate gene sequence variant discovery projects for instance, targeted MPS provides an alternative to the more costly, time-consuming, and lower-throughput approaches previously used in large-scale case-control mutation screening studies (Citation3, Citation4). Familial genetic testing for high penetrance cancer genes, such as BRCA1 and BRCA2, has also benefited from the significant cost reduction offered by targeted MPS (Citation5).
Targeted MPS methods are generally based on PCR amplification or hybridization capture approaches (Citation6–8). Commonly used performance measures for these methods are: (i) percentage of target bases represented by one or more sequence reads (coverage); (ii) percentage of sequences that map to the intended target (on-target); (iii) variability in sequence coverage across target regions (uniformity); (iv) cost; (v) ease of use; and (vi) amount of input DNA required per experiment or per megabase of target. Although unanimously recognized as a highly sensitive, specific, and uniform approach for targeted MPS, PCR-based MPS in its current form also has limitations relating to cost, throughput, and the ability to multiplex to a useful degree. Simultaneous production of many amplicons can lead to differential production of amplicons or nonspecific or failed amplification. Additionally, many PCR-MPS methods require labor-intensive normalization to achieve equimolar pooling of separate PCR products prior to sequencing. Commercial solutions have been developed by several manufacturers recently for library building based on high-multiplex PCR (high-plex PCR), for example, Ion Ampliseq (Life Technologies) (Citation9), TruSeq Amplicon (Illumina), and Haloplex (Agilent) (Citation10). These systems have different constraints in terms of assay design, cost, input material requirements, and turn-around times. Commercial systems have also been developed for miniaturized PCR by microfluidics. The Access Array system (Fluidigm) allows 48 single-plex assays across 48 samples with a relatively modest quantity of input template (50 ng/sample), and the RainStorm technology (RainDance Technologies) is based on the generation of microdroplets in an oil emulsion (Citation11). This approach allows ∼4,000 simultaneous amplifications, but its limitations include the requirement for a very high amount of input DNA and sequential processing of individual samples. Microfluidic chips and their associated instrumentation are prohibitively expensive for small research or diagnostic labs. The current approaches each have their own advantages and limitations, and the method of choice is thus dependent on the application, required performance, input material, ease of use, cost, and available instrumentation.
Use of MPS in diagnostic settings also presents specific technical challenges. For example, methods for sequencing DNA extracted from formalin-fixed, paraffin-embedded (FFPE) material have had variable success so far (Citation12). There is great potential for the use of FFPE specimens in cancer studies and clinical diagnostics because pathology departments routinely collect and store them.
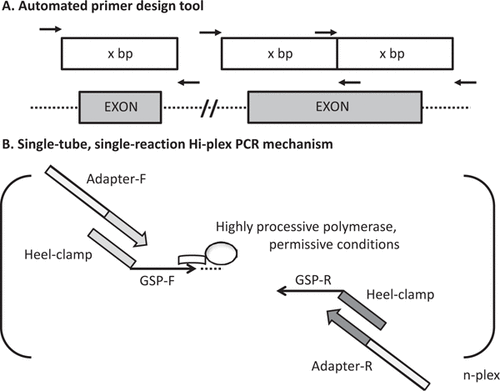
To address the limitations described above, we have developed an in-house-customizable PCR-MPS technology that we call Hi-Plex (). Hi-Plex panels can be designed and customized in minutes using our simple automated primer design software. Preparation for genetic testing or screening (library building) is performed via a single PCR amplification, followed by size selection, for a fraction of the reagent cost and hands-on time required by other methods. We have tested Hi-Plex in a 60-plex assay using all of the protein-coding and some of the 5′-untranslated and 3′-untranslated regions of the breast cancer predisposition genes PALB2 and XRCC2 (Citation13–15), both on cell line-derived genomic DNA and FFPE tumor-derived DNA.
A) Gene-specific primer regions are designed to yield product with a defined size (100 bp) within a narrow window to allow stringent size selection. B) In a single reaction, 5′ heeled (heel clamp) gene-specific primers (GSP-F and GSP-R) representing all targeted amplicons (n-plex) are combined with adapter primers for PCR-based thermocycling. A highly processive and high-fidelity thermostable DNA polymerase (e.g., Phusion high-fidelity polymerase) is used, along with permissive reaction conditions for annealing and extension. This experimental design includes GSPs of 20 to 30 bases, forward heel clamp comprising 23 bases, and reverse heel clamp of 19 bases. The forward and reverse adapter primers were 41 and 30 bases, respectively (see Supplementary Table S1). It is envisaged that a variety of heeled primer designs with various element attributes would prove effective.
Materials and methods
For our primer software named Hiplex-primer (publicly available for download as a set of Python scripts from https://github.com/bjpop/hiplex-primer), we input the exon coordinates for PALB2 and XRCC2, set the length of the primer intervening sequence to 100 bases, and set a target melting temperature for gene-specific primer regions at 64°C, along with a maximum length of 30 bases and an allowable gene-specific region length variability of 10 bases. Each of these parameters can be modified by the user. To the gene-specific primer region outputs, we added 5′ heel sequences corresponding to adapters that are compatible with Ion Torrent (Life Technologies, Foster City, CA) sequencing chemistry ( and Supplementary Table S1). For our experiments, no primers were altered from the initial automated design and all gene-specific primers were present in equal concentration. All oligonucleotide sequences are listed in Supplementary Table S1 and were synthesized by Integrated DNA Technologies (Coralville, IA). Adapter primers were HPLC purified, but all other primers were supplied as standard desalting grade.
Input material consisted of genomic DNA derived from an Epstein Barr Virus-transformed lymphoblastoid cell line (LCL) generated in house, and from FFPE breast cancer tumor tissue collected in 1997 as part of the Australian Breast Cancer Family Study (ABCFS) (Citation16). Extraction was performed using the QIAmp DNA Blood Kit (Qiagen, Dusseldorf, Germany) and QIAamp DNA FFPE Tissue Kit, respectively. DNA was quantitated using the Qubit dsDNA Assay system (Life Technologies).
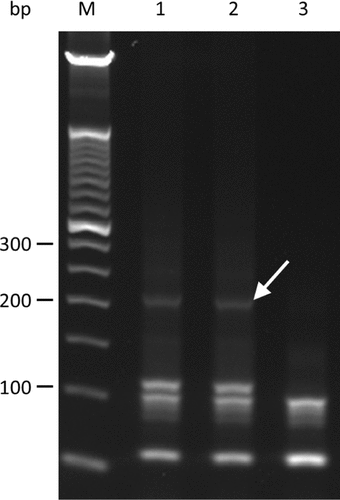
A 50 µL PCR reaction was made up of 1× Phusion HF PCR buffer (ThermoScientific, Waltham, MA), 2 U of Phusion Hot Start II High-Fidelity DNA Polymerase, 400 µM dNTPs (Bioline, London, UK), 0.5 µM gene-specific primer pool (0.004 µM each individual gene-specific primer), 2.5 mM MgCl2, and either 100 or 25 ng input genomic DNA. The following steps were used for the PCR: 98°C for 1 min, 6 cycles of [98°C for 30 s, 50°C for 1 min, 55°C for 1 min, 60°C for 1 min, 65°C for 1min, 70°C for 1 min], addition of 2 µL of a mix of 50 µM IT_P1_noT and IT-A-key primers (to achieve a final reaction concentration of 2 µM for each), then a further 19 cycles of [98°C for 30 s, 50°C for 1 min, 55°C for 1 min, 60°C for 1 min, 65°C for 1 min, 70°C for 1 min], followed by incubation at 60°C for 20 min. Eight µl of product was subjected to 1.5% agarose/ TBE (w/v) gel electrophoresis. The approximately 220 bp band containing our target library was excised and the DNA was extracted and purified using the Qiagen QIAEX II kit (Qiagen). shows an agarose gel profile for the Hi-Plex product.
M indicates 50 bp ladder (Life Technologies). Lanes 1 to 3 were loaded with 1.5 µL Hi-Plex products after applying the thermocycling conditions described in the main text. Hi-Plex reactions for the products loaded in lanes 1 and 2 contained genomic DNA template, whereas the reaction for the product loaded in lane 3 had the equivalent template volume substituted with water (no DNA control). The arrow indicates the library band.
Hi-Plex of the 100 ng LCL-derived DNA was conducted using the Ion 314 chip/Ion PGM 200 Sequencing Kit and mapped using Torrent Suite v.3.4.1 (Life Technologies). Hi-Plex with 100 ng or 25 ng FFPE-derived DNA used the Ion 316 chip/Ion PGM 200 Sequencing Kit and Torrent Suite v3.4.2. In order to assess individual amplicon PCR efficiencies, read depths were determined at the midpoints of the sequences between the gene-specific primer regions.
Results and discussion
We have developed our Hiplex-primer software to implement our automated approach to primer design. This software tool accepts target coordinates and the primer intervening sequence size as user inputs. The user also specifies an intended length for gene-specific primer regions and a maximum gene-specific primer region length. The software assesses primer melting temperatures based on the simplified assumption that each G or C contributes 4°C to the total melting temperature and each A or T contributes 2°C (the ‘4 and 2’ rule). The software searches in and around these coordinate blocks to minimize the differences between predicted melting temperatures for gene-specific primer sequences and a user-defined target melting temperature.
Relative amplification bias is restricted in Hi-Plex by a combination of mechanisms. Use of 5′ heel clamps, which have adapter sequences that are used subsequently by the sequencing chemistry, can reduce amplification bias (Citation17). Adapter primers are added at the early stages of thermocycling. For the majority of amplification cycles, successful priming is not dependent on gene-specific primers; rather, priming of all targets in the pool can be driven by the same two adapter primers. Generally, smaller targets are more efficiently amplified than larger ones (Citation18, Citation19). Hi-Plex defines amplicon sizes within a narrow size range, thus eliminating size-related bias. Our method also uses a highly processive DNA polymerase, preferably one with high fidelity (e.g., Phusion Hot Start II High-Fidelity DNA Polymerase), and permissive thermocycling conditions. The system can afford to use relatively low temperature annealing conditions because the size selection step that follows PCR eliminates the great majority of off-target reaction by-products. As such, Hi-Plex tolerates a broad range of primer types with different G/C-contents and actual primer annealing temperatures. In addition to permissive annealing, Hi-Plex uses permissive extension conditions Previous studies have demonstrated that low G/C-content amplicons can benefit from relatively low extension temperatures (Citation20). On the other hand, sequences with relatively high G/C-content can benefit from relatively high temperature thermocycling conditions (Citation21). Our preferred approach to allow for successful amplification of all amplicons representing a broad spectrum of primer and intervening sequence contexts is to apply a gradient of annealing/extension temperatures during thermocycling. Hi-Plex uses relatively long cycle steps to allow greater opportunity for priming and complete extension. In combination, these design elements free the system from many sequence contextual design constraints.
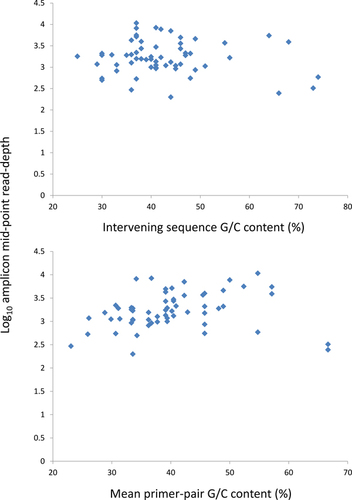
Application of Hi-Plex to 100 ng LCL-derived DNA showed that 93.33% (56/60), 98.33% (59/60), and 100% of targeted amplicons were represented within 5-fold, 10-fold, and 12.5-fold of the mean on-target coverage, respectively. When mapped to the whole human genome, 86.94% of Hi-Plex reads were aligned within PALB2 and XRCC2, with a total number of on-target reads and a mean number of on-target reads per amplicon of 147,838 and 2463.96, respectively (314 chip). The number of on-target reads ranged from 199 (12.38-fold less than the mean) to 10,746 (4.36-fold higher than the mean). illustrates the relative representation of the 60 amplicons in relation to mean primer pair G/C-content and intervening sequence G/C-content. The G/C-content of the gene-specific regions of individual primers ranged from 10.35% to 66.67%. The G/C-content of the intervening sequences ranged from 25% to 74% and amplicons included G/C-rich 5′ untranslated regions. It is worth noting that the single amplicon that yielded a number of on-target reads more than 10-fold from the mean (12.38- fold lower) included a primer in which the 14 3′-most nucleotides had a G/C-content of only 14.29% and included a predicted perfect 4 bp hairpin structure. The LCL was derived from a known heterozygous carrier of the pathogenic PALB2 c.3113 G > A mutation. Of the 1095 reads at this position, 552 (50.41%) and 542 (49.50%) represented the major and minor alleles, respectively. This supports the value of Hi-Plex for gene screening projects, as the approach can accurately detect mutations.
When applied to 100 ng FFPE-derived DNA, Hi-Plex resulted in 78.33% (47/60), 91.67% (55/60), and 100% of targeted amplicons represented within 5-fold, 10-fold, and 25-fold of the mean. A total of 2,556,204 reads (97.33%) were on-target, with a mean of 42,603.4 on-target reads per amplicon. The number of on-target reads ranged from 2056 (20.72-fold less than the mean) to 237,945 (5.59-fold higher than the mean) (316 chip).
When the amount of input FFPE-derived DNA was reduced to 25 ng, Hi-Plex resulted in 90% (54/60), 98.33% (59/60), and 100% of targeted amplicons represented within 5-fold, 10-fold, and 12.5 fold of the mean. In this assay, a total of 2,454,875 reads (95.14%) were on-target, with a mean of 40,914.58 on-target reads per amplicon. The number of on-target reads ranged from 4046 (10.11-fold less than the mean) to 235,904 (5.77-fold higher than the mean) (316 chip). Supplementary Figures S1 and S2 show the relative representation of the 60 amplicons in relation to mean primer pair G/C-content and intervening sequence G/C-content observed in the 100 ng and 25 ng FFPE tumor-derived DNA Hi-Plex experiments. The lowest performer in both FFPE runs was the amplicon with the highest primer-intervening sequence G/C-content (74%).
This study has demonstrated the advantages of the Hi-Plex method using Ion Torrent sequencing chemistry. Our system showed minimal amplification biases associated with differential primer efficiencies and eliminates the need for extensive primer concentration optimization, primer redesign, or normalization/pooling of multiple separate PCR products to achieve uniform coverage. We have shown the benefits of Hi-Plex in the context of degraded FFPE specimens, along with low levels of input DNA, without compromising sequencing data quality. That Hi-Plex yielded the narrowest range of relative amplicon representation using 25 ng FFPE-derived material suggests that fragmented template might reduce the potential for off-target amplicons to compete with on-target amplicons and that the system may perform better using relatively low inputs of such material. The use of newer versions of chemistry and software could also have contributed to the improved performance compared with the assay of LCL-derived DNA.
Existing commercial solutions for PCR-based MPS proposed by Life Technologies (Ion Ampliseq), Illumina (TruSeq Amplicon), and Agilent (Haloplex) require expensive reagents, including multiple enzyme formulations and highly specialized oligonucleotide mixtures that are difficult to produce. They also involve laborious protocols with many enzymatic processing and purification steps, and can be limited in the scope of targetable regions. They typically require two days of library preparation involving many hands-on steps, with each step increasing the potential for error, such as the introduction of PCR contamination. Haloplex currently costs in excess of $10,000 AUD (Australian dollars; >$9000 USD) to prepare 96 specimen libraries and is particularly constrained in terms of design due to the need for fortuitous restriction enzyme sites and other local nucleotide sequence. Requirements for particular sequence contexts also constrain the other chemistries, e.g., the Life Technologies design software only allowed ∼30% of XRCC2 to be targeted. AmpliSeq is restricted to use with the relatively error-prone Ion Torrent sequencing chemistry that exhibits ∼98.2% per base accuracy compared with >99.6% for Illumina sequencing chemistry (Citation22). None of these approaches allow fine control of the product size range, which has implications for relative amplification efficiencies of amplicons and the potential to apply stringent sequencing artifact filtering approaches via paired-end read comparison (Citation6).
Because Hi-Plex performed so well across a wide variety of amplicons without alteration of gene-specific primer sequences or relative concentrations, we envisage that these methods will be broadly applicable across a wide range of targets with minimal optimization. Should fine-tuning be required for given applications, however, redesign of under-performing primer pairs or adjustment of primer concentrations for outlier amplicons remain viable options. Currently, our primer design software implements very basic algorithms. It is likely that future improvements can be made by such measures as taking account of primer secondary structure predictions and avoiding low G/C-content toward the 3′ end of gene-specific primers. Adoption of more sophisticated melting temperature prediction algorithms, such as including nearest-neighbor effects, might confer further benefits (Citation23). More sophisticated existing programs for multiplex PCR primer design do not allow precise definition of the amplicon size, however (Citation24–26). Hi-Plex has been designed so that genes or genomic regions could easily be swapped in and out of a panel, without extensive protocol optimization or primer redesign.
In future experiments, we will test Hi-Plex for considerably higher parallelization, with the aim of achieving robust thousands-plex single-tube multiplexing. The mechanisms underlying Hi-Plex suggest that this should be possible without extensive protocol adjustment. Theoretically, Hi-Plex should be sequencing platform agnostic, able to be transferred across chemistries simply by swapping adapter sequences. The ability to define amplicon size using Hi-Plex should also allow stringent sequencing chemistry artifact-filtering using completely overlapping paired-end reads. When paired reads show discrepancy at given positions, confidence in the call can be assigned more appropriately than would otherwise be possible.
The size selection step could optionally be automated using a system such as the Pippin Prep System (Sage Science). It should not be necessary to conduct separate size selection steps for separate Hi-Plex libraries. Use of specimen encoding, for example barcoded adapters, should allow a large number of Hi-Plex PCR products to be size selected at once. The reagent costs for the Hi-Plex library build (primers, PCR components, and gel extraction), assuming high-throughput, are around one dollar per specimen, rendering this approach orders of magnitude more cost-effective than the alternatives.
Having been successfully demonstrated on both cell line- and FFPE-derived material, Hi-Plex represents an exciting method for a range of molecular screening applications, including diagnostics, disease predisposition profiling, and disease gene discovery.
Competing interests
The authors declare no competing interests.
A high-plex PCR approach for massively parallel sequencing
Download PDF (144.4 KB)Acknowledgments
This work was supported by the Australian National Health and Medical Research Council (NHMRC) (APP1025879 and APP1029974), by a Victorian Life Sciences Computation Initiative (VLSCI) grant number VR0182 on its Peak Computing Facility at the University of Melbourne, an initiative of the Victorian Government, and by the Victorian Breast Cancer Research Consortium (VBCRC). TN-D is a Susan G. Komen for the Cure Postdoctoral Fellow. MCS is a VBCRC Group Leader and Senior Research Fellow of the NHMRC. We thank the Australian Breast Cancer Family Study (ABCFS, Principal Investigator John Hopper) for providing the cell-line derived DNA, and Ee Ming Wong for providing the FFPE tumor-derived DNA.
Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at: www.tandfonline.com/doi/suppl/10.2144/000114052
Additional information
Funding
References
- Mardis, E.R. 2008. The impact of next-generation sequencing technology on genetics.Trends Genet.24:133–141.
- Shendure, J. and H.Ji. 2008. Next-generation DNA sequencing.Nat. Biotechnol.26:1135–1145.
- Tavtigian, S.V., P.J.Oefner, D.Babikyan, A.Hartmann, S.Healey, F.Le Calvez-Kelm, F.Lesueur, G.B.Byrnes, et al.. 2009. Rare, evolutionarily unlikely missense substitutions in ATM confer increased risk of breast cancer.Am. J. Hum. Genet.85:427–446.
- Le Calvez-Kelm, F., F.Lesueur, F.Damiola, M.Vallee, C.Voegele, D.Babikyan, G.Durand, N.Forey, et al.. 2011. Rare, evolutionarily unlikely missense substitutions in CHEK2 contribute to breast cancer susceptibility: results from a breast cancer family registry case-control mutation-screening study.Breast Cancer Res.13:R6.
- Morgan, J.E., I.M.Carr, E.Sheridan, C.E.Chu, B.Hayward, N.Camm, H.A.Lindsay, C.J.Mattocks, et al.. 2010. Genetic diagnosis of familial breast cancer using clonal sequencing.Hum. Mutat.31:484–491.
- Meldrum, C., M.A.Doyle, and R.W.Tothill. 2011. Next-generation sequencing for cancer diagnostics: a practical perspective.Clin. Biochem. Rev.32:177–195.
- Moorthie, S., C.J.Mattocks, and C.F.Wright. 2011. Review of massively parallel DNA sequencing technologies.The HUGO journal5:1–12.
- Mamanova, L., A.J.Coffey, C.E.Scott, I.Kozarewa, E.H.Turner, A.Kumar, E.Howard, J.Shendure, and D.J.Turner. 2010. Target-enrichment strategies for next-generation sequencing.Nat. Methods7:111–118.
- Yang, M.M., A.Singhal, S.R.Rassekh, S.Yip, P.Eydoux, and C.Dunham. 2012. Possible differentiation of cerebral glioblastoma into pleomorphic xanthoastrocytoma: an unusual case in an infant.Journal of neurosurgery. Pediatrics9:517–523.
- Schulz, E., A.Valentin, P.Ulz, C.Beham-Schmid, K.Lind, V.Rupp, H.Lackner, A.Wolfler, et al.. 2012. Germline mutations in the DNA damage response genes BRCA1, BRCA2, BARD1 and TP53 in patients with therapy related myeloid neoplasms.J. Med. Genet.49:422–428.
- Tewhey, R., J.B.Warner, M.Nakano, B.Libby, M.Medkova, P.H.David, S.K.Kotsopoulos, M.L.Samuels, et al.. 2009. Microdroplet-based PCR enrichment for large-scale targeted sequencing.Nat. Biotechnol.27:1025–1031.
- Schweiger, M.R., M.Kerick, B.Timmermann, M.W.Albrecht, T.Borodina, D.Parkhomchuk, K.Zatloukal, and H.Lehrach. 2009. Genome-wide massively parallel sequencing of formaldehyde fixed-paraffin embedded (FFPE) tumor tissues for copy-number- and mutation-analysis.PLoS ONE4:e5548.
- Rahman, N., S.Seal, D.Thompson, P.Kelly, A.Renwick, A.Elliott, S.Reid, K.Spanova, et al.. 2007. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene.Nat. Genet.39:165–167.
- Southey, M.C., Z.L.Teo, J.G.Dowty, F.A.Odefrey, D.J.Park, M.Tischkowitz, N.Sabbaghian, C.Apicella, et al.. 2010. A PALB2 mutation associated with high risk of breast cancer.Breast Cancer Res.12:R109.
- Park, D.J., F.Lesueur, T.Nguyen-Dumont, M.Pertesi, F.Odefrey, F.Hammet, S.L.Neuhausen, E.M.John, et al.. 2012. Rare mutations in XRCC2 increase the risk of breast cancer.Am. J. Hum. Genet.90:734–739.
- Hopper, J.L., G.Chenevix-Trench, D.J.Jolley, G.S.Dite, M.A.Jenkins, D.J.Venter, M.R.McCredie, and G.G.Giles. 1999. Design and analysis issues in a population-based, case-control-family study of the genetic epidemiology of breast cancer and the Co-operative Family Registry for Breast Cancer Studies (CFRBCS).J. Natl. Cancer Inst. Monogr.25:95–100.
- Rose, T.M., J.G.Henikoff, and S.Henikoff. 2003. CODEHOP (COnsensus-DEgenerate Hybrid Oligonucleotide Primer) PCR primer design.Nucleic Acids Res.31:3763–3766.
- Wong, M.L. and J.F.Medrano. 2005. Real-time PCR for mRNA quantitation.BioTechniques39:75–85.
- Yuan, J.S., A.Reed, F.Chen, C.N.Stewart, Jr. 2006. Statistical analysis of real-time PCR data.BMC Bioinformatics7:85.
- Su, X.Z., Y.Wu, C.D.Sifri, and T.E.Wellems. 1996. Reduced extension temperatures required for PCR amplification of extremely A+T-rich DNA.Nucleic Acids Res.24:1574–1575.
- Aird, D., M.G.Ross, W.S.Chen, M.Danielsson, T.Fennell, C.Russ, D.B.Jaffe, C.Nusbaum, and A.Gnirke. 2011. Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries.Genome Biol.12:R18.
- Quail, M.A., M.Smith, P.Coupland, T.D.Otto, S.R.Harris, T.R.Connor, A.Bertoni, H.P.Swerdlow, and Y.Gu. 2012. A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers.BMC Genomics13:341.
- Kibbe, W.A. 2007. OligoCalc: an online ol igonucleotide properties calculator.Nucleic Acids Res.35:W43–W46.
- Holleley, C.E. and P.G.Geerts. 2009. Multiplex Manager 1.0: a cross-platform computer program that plans and optimizes multiplex PCR.BioTechniques46:511–517.
- Shen, Z., W.Qu, W.Wang, Y.Lu, Y.Wu, Z.Li, X.Hang, X.Wang, et al.. 2010. MPprimer: a program for reliable multiplex PCR primer design.BMC Bioinformatics11:143.
- Souvenir, R., J.Buhler, G.Stormo, and W.Zhang. 2007. An iterative method for selecting degenerate multiplex PCR primers.Methods Mol. Biol.402:245–268.

