
Abstract
Tools for studying replication fork dynamics are critical for dissecting the mechanisms of DNA replication, DNA repair, histone deposition, and epigenetic memory. Isolation of protein on nascent DNA (iPOND) is an elegant method for purifying replication fork proteins. Here, we present accelerated native iPOND (aniPOND), a simplification of the iPOND procedure with improved protein yield. Cell membrane lysis and nuclei harvesting are combined in one step to reduce washes and minimize sample loss. A mild nuclei lysis protocol is then used to better preserve DNA-protein complexes. aniPOND is faster than iPOND, avoids formaldehyde cross-linking, and improves protein yield 5- and 20-fold for the CAF1-complex or PCNA respectively. Moreover, using aniPOND, but not iPOND, we could detect the polycomb repressive complex 2 (PRC2) components SUZ12, EZH2, and RBBP4 at replication forks. This faster, higher-yield method will facilitate MS analysis of replication fork complexes.
Catching more proteins from the POND
Analyzing the dynamics of replication fork progression is critical for understanding DNA replication and repair as well as epigenetic regulation involving the deposition of histones and other chromatin proteins. Recently, isolation of protein on nascent DNA (iPOND) was developed to purify proteins found at the replication fork. Nascent DNA at the replication fork is labeled through the incorporation of a brief pulse of the thymidine analog 5-ethynyl-2’-deoxyuridine (EdU), followed by formaldehyde crosslinking of chromatin proteins to DNA and the covalent linkage of biotin azide to the alkyne group of EdU through a copper-catalyzed “click” reaction. After sonication of the chromatin, biotinylated nascent DNA and associated proteins are isolated using streptavidin-coated beads and proteins are analyzed by Western blotting after the reversal of the formaldehyde crosslinking. iPOND, however, has drawbacks, including modest protein yields and the need for formaldehyde crosslinking that can affect Western blotting and mass spectrometry protein analysis. In this issue, R. Bremner and colleagues at the University of Toronto (Ontario, Canada) describe their faster and simpler adaptation of iPOND, which they call accelerated native iPOND (aniPOND). aniPOND allows for the isolation of proteins under native conditions with a 5- to 20-fold improvement in yield. After initial EdU incorporation, a key modification in aniPOND is to combine the cell harvesting, lysis, extraction of soluble cytoplasmic and nuclear proteins, and collection of nuclei all into a single step by adding nuclei extraction buffer directly to cells in the flask. This reduction in sample manipulation likely accounts for the enhanced protein recovery observed when using aniPOND. After the click reaction, chromatin is sonicated in a 1% NP40 buffer instead of the SDS buffer used for iPOND to maintain the protein complexes in their native state. Two single sonication/centrifugation steps are then performed to remove non-chromatin proteins, followed by extensive sonication to thoroughly shear and solubilize the chromatin. Isolation of the biotin-labeled replication forks using streptavidin beads is done as in iPOND, except the crosslinking reversal step is eliminated since aniPOND does not utilize formaldehyde crosslinking. Using aniPOND, the authors demonstrate increased capture of abundant replication fork proteins as well as detection of rarer proteins.
See “A rapid and efficient method to purify proteins at replication forks under native conditions” on page 204.
We developed aniPOND to capture proteins at replication forks with improved efficiency under native conditions. Compared with the original approach, aniPOND increased protein yield 5–20 fold.
iPOND (isolation of protein on nascent DNA) was developed by Sirbu et al. in 2011 (Citation1) to study DNA replication. A similar procedure was presented in the same year by Kliszczak et al. (Citation2). First, nascent DNA is labeled by a brief pulse of the thymidine analog 5-ethynyl-2′- deoxyuridine (EdU) (Citation3). Then, biotin azide is covalently linked to the alkyne functional group on EdU in the presence of copper catalyst via a click reaction (Citation4). Finally, biotinylated DNA is precipitated using streptavidin-coated beads and Western blotting is used to identify the associated proteins. Here, we build on this elegant method to generate a new protocol that is simpler, faster, and significantly improves protein yield (see Supplementary Material for a detailed protocol).
Limitations of iPOND include modest protein yield and the use of formaldehyde cross-linking to preserve DNA-protein complexes. Formaldehyde cross-linking could interfere with protein identification by mass spectrometry (MS) and detection of large molecular weight proteins by Western blotting if not fully reversed (Citation5,Citation6). In 2012, Sirbu et al. (Citation6) proposed an alternate protocol, native iPOND (niPOND), for performing iPOND in the absence of formaldehyde, although results were not presented.
We were able to capture replication fork proteins using iPOND, but not with niPOND (data not shown). Here, we present accelerated native iPOND (aniPOND), which improves capture efficiency on average by an order of magnitude compared to iPOND and cuts the total time needed to perform the assay in half compared to niPOND () (Supplementary Material).
(A) Flow-chart showing an overview of the iPOND (blue), niPOND (red), and aniPOND (magenta) procedures. O/N = overnight. Total or hands-on time for each protocol is listed at the bottom of the flowchart. (B) Representative Western blot of input (I) or captured (C) proteins following iPOND or aniPOND on SW13 cells labeled for 10 min with EdU. (C) Quantification of Western blot results +/- SD (n = 4). Statistics were performed using Student’s t-test, * P < 0.005 and ** P < 0.001. In all experiments, a total of ∼6x107 cells in 4 175 cm2 tissue flasks was used. Cells were seeded at 1.5x107 cells per flask. Input = 0.25% cell equivalent (1.5x105) cells. Capture = 25% cell equivalent (1.5x107) cells.
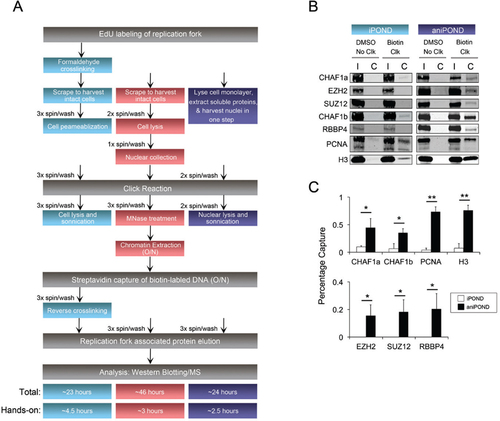
In the iPOND and niPOND protocols, cells are labeled with EdU, scraped, spun, and washed, and then permeabilized or lysed prior to the click reaction (). For aniPOND, harvesting and lysis were performed in the flask simultaneously. Nuclear extraction buffer (NEB) was used to extract most of the soluble cytoplasmic and nuclear proteins and generate nuclei in one step (). Chromatin bound proteins were retained in the nuclear fraction (Supplementary Figure S1). This single-step procedure reduces manipulation of the sample, likely contributing to the enhanced recovery with aniPOND.
After a one-hour click reaction, biotinylated chromatin needs to be sheared and extracted from nuclei for the subsequent streptavidin capture step (). In the original iPOND protocol, chromatin is sonicated in a buffer containing the strong ionic detergent sodium dodecyl sulfate (SDS). This approach cannot be used in native preps as SDS or milder ionic detergents such as sodium deoxycholate disrupt protein complexes. The niPOND protocol suggests the use of micrococcal nuclease to digest chromatin, followed by overnight extraction in buffer containing 0.1% Triton-X. Using this approach we recovered <3 mg of protein from 6x107 cells. In our new aniPOND approach, we first performed two single sonication/spin cycles on nuclei in a buffer with 1% NP40 to release more non-chromatin protein, then applied extensive sonication to shear and solubilize the chromatin in the remaining sample. This strategy yielded DNA fragments of ∼150 bp (Protocol ) and >7 mg of protein. The niPOND approach requires an overnight incubation, but the new aniPOND version of this step is complete in one hour.
Following chromatin extraction, the next step is to purify biotin labeled replication forks using streptavidin (). The protocols for iPOND, niPOND, and aniPOND are similar at this stage, except that iPOND requires an extra cross-linking reversal step. We ran Western blots to compare the capture efficiency of iPOND and aniPOND; niPOND data was not included because that technique was unsuccessful in our hands. Relative to iPOND, aniPOND significantly increased the yield of the CHAF1a and CHAF1b subunits of the CAF1 histone chaperone complex by 5-fold, and PCNA by 20-fold (,). aniPOND not only improved the capture of these abundant replication fork proteins, but also allowed detection of rarer proteins such as PRC2 subunits (,). A recent study using iPOND followed by MS described two new replication fork proteins, TFII-I and ZNF24 (Citation7). We also detected these factors using aniPOND (Supplementary Figure S3). The replisome component MCM-3 was also recovered by aniPOND (Supplementary Figure S3). In addition, aniPOND was effective when performed on a different cell line (Supplementary Figure S2).
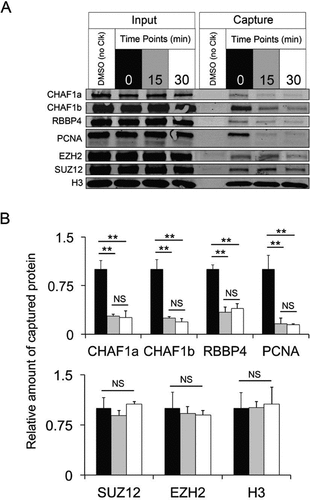
We also tested aniPOND in a more complex pulse-chase experiment. Similar to the above data, aniPOND captured replication fork proteins (PCNA and CAF1 complex) after a 10 min EdU pulse, and a subsequent 15 or 30 min chase period in which thymidine displaced replication forks from EdU-labeled DNA, diminishing the levels of PCNA and CAF1 complex ( and ). PRC2 subunits and histone H3 levels remained constant ( and ), indicating that these proteins remain on nascent DNA even after the replication fork passes. These observations are in agreement with published data (Citation1,Citation8).
(A) Western blot of input and captured proteins following aniPOND on SW13 cells incubated with EdU for 10 min and then chased in thymidine for the indicated times. (B) Quantification of Western blots +/- SD (n = 3). Statistics were performed with Student’s t-test, ** P < 0.001, NS = not significant. The amount of capture at 15 and 30 min is normalized to the average at time 0. The amount of cells used and protein loaded was as in .
In summary, aniPOND provides a simpler, faster method to purify replication fork proteins under native conditions with higher yield. Coupled with MS, our approach may expand the list of replication fork proteins, given that it captures less abundant proteins such as members of the PRC2 complex. Comprehensive MS analyses will be required to determine whether aniPOND consistently detects more proteins than iPOND.
Author contributions
K.H.T.L. performed all experiments. K.H.T.L., M.A.E.H. and R.B. developed the protocol. K.H.T.L. and R.B. wrote the manuscript.
Competing interests
The authors declared no competing financial interests.
Additional File 1
Download Zip (607.8 KB)Acknowledgments
This work was supported by the Canadian Cancer Society Research Institute (CCSRI) and the Krembil Foundation.
Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at: www.tandfonline.com/doi/suppl/10.2144/000114089
References
- Sirbu, B.M., F.B.Couch, J.T.Feigerle, S.Bhaskara, S.W.Hiebert, and D.Cortez. 2011. Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev.25:1320–1327.
- Kliszczak, A.E., M.D.Rainey, B.Harhen, F.M.Boisvert, and C.Santocanale. 2011. DNA mediated chromatin pull-down for the study of chromatin replication. Sci Rep.1:95.
- Salic, A. and T.J.Mitchison. 2008. A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc. Natl. Acad. Sci. USA105:2415–2420.
- Moses, J.E. and A.D.Moorhouse. 2007. The growing applications of click chemistry. Chem. Soc. Rev.36:1249–1262.
- Toews, J., J.C.Rogalski, T.J.Clark, and J.Kast. 2008. Mass spectrometric identification of formaldehyde-induced pept ide modificat ions under in vivo protein cross-linking conditions. Anal. Chim. Acta618:168–183.
- Sirbu, B.M., F.B.Couch, and D.Cortez. 2012. Monitoring the spatiotemporal dynamics of proteins at replication forks and in assembled chromatin using isolation of proteins on nascent DNA. Nat. Protoc.7:594–605.
- Lopez-Contreras, A.J., I.Ruppen, M.Nieto-Soler, M.Murga, S.Rodriguez- Acebes, S.Remeseiro, S.Rodrigo-Perez, A.M.Rojas, et al.. 2013. A Proteomic Characterization of Factors Enriched at Nascent DNA Molecules. Cell Rep.3:1105–1116.
- Petruk, S., Y.Sedkov, D.M.Johnston, J.W.Hodgson, K.L.Black, S.K.Kovermann, S.Beck, E.Canaani, et al.. 2012. TrxG and PcG Proteins but Not Methylated Histones Remain Associated with DNA through Replication. Cell150:922–933.

