
Abstract
A large number of methods are available to deplete ribosomal RNA reads from high-throughput RNA sequencing experiments. Such methods are critical for sequencing Drosophila small RNAs between 20 and 30 nucleotides because size selection is not typically sufficient to exclude the highly abundant class of 30 nucleotide 2S rRNA. Here we demonstrate that pre-annealing terminator oligos complimentary to Drosophila 2S rRNA prior to 5′ adapter ligation and reverse transcription efficiently depletes 2S rRNA sequences from the sequencing reaction in a simple and inexpensive way. This depletion is highly specific and is achieved with minimal perturbation of miRNA and piRNA profiles.
Enriching your RNA-seq efforts
When it comes to any next-generation sequencing experiment, scientists want to maximize the number of informative reads. In the case of high-throughput RNA sequencing (RNA-seq), this means eliminating those RNA species that are not of interest for the experiment. The trouble is that, in the RNA world, those RNAs that are not of direct interest are often the ones that are most abundant. A number of techniques have been developed to enrich for specific classes of RNA prior to RNA-seq, including methods based on the presence of poly-A tails, size selection, or hybridization to specific DNA oligonucleotides. While these approaches have proven effective in many cases, problems can still arise. Take for instance the case of Drosophila small RNA sequencing (miRNA, piRNA, siRNA). Here, the RNAs of interest are 20–30 nucleotides in length, but so too is the highly abundant class of 2S rRNA, making small RNA enrichment in this species a particular challenge. But in a Benchmark article in this issue of BioTechniques, Michelle Wickersheim and Justin Blumenstiel from the University of Kansas present a clever solution to enriching for small RNAs—one that does not require additional hybridization or size selection techniques. Wickersheim and Blumenstiel hypothesized that 2S rRNA could be most effectively depleted during the ligation and cDNA synthesis steps of sequencing library construction by not allowing these RNAs to take part in the reactions. The authors accomplished this depletion using terminator blocking oligonucleotides specific for 2S rRNAs that annealed to the templates prior to the 5′ adapter ligation step. In this way, only small RNAs of interest should be reverse transcribed, barcoded, and subsequently sequenced. When Wickersheim and Blumenstiel tested their terminator oligo blocking methodology, they found less than 0.1% of total reads obtained following depletion using blocking oligos were from 2S rRNA. Although the method was demonstrated for Drosophila small RNAs, this approach will likely find favor with all scientists interested in enriching their target small RNA populations.
See “Terminator oligo blocking efficiently eliminates rRNA from Drosophila small RNA sequencing libraries” on page 269.
Keywords: :
Here we present a simple method for eliminating 2S RNA from small RNA sequencing reactions in Drosophila with significant time and cost saving. This method may also have broader applications for other forms of RNA-seq.
High-throughput RNA sequencing (RNA-seq) has revolutionized our understanding of RNA biology, but the presence of high abundance RNA species that are not of direct interest can be problematic. For this reason, there is great interest in developing methods that enrich specific classes of RNA for RNA-seq applications. Historically, the most commonly used method for enriching mRNA depends on the presence of poly(A) tails. However, this method is not applicable in non-eukaryotes lacking polyadenylation; it is also incapable of enriching for many non-coding RNAs and degraded RNAs that lack poly(A) tails. In contrast to enriching for poly(A), an alternative approach is to directly deplete nuisance classes of RNA. A recent survey (Citation1) compared different methods (Citation2–6) for depleting nuisance species of rRNA, favoring rRNA hybridization to DNA oligos and removal of RNA-DNA hybrids by RNase H treatment.
For sequencing small RNAs (miRNAs, siRNAs and piRNAs), enrichment is usually achieved by size selection. Since these classes of RNA are typically between 20 and 30 nt, size selection from total RNA, followed by ligation, PCR, and further size selection, generally achieves good results (see Reference 7 for a review of small RNA sequencing methods). In Drosophila, however, size selection is not typically sufficient because the highly abundant class of 2S rRNA is 30 nt long. To deal with this problem, two approaches have been developed. One method, using RNase H, employs hybridization with DNA oligos complementary to 2S rRNA and directed degradation (Citation8). A second method depletes 2S rRNA by hybridization using oligos bound to magnetic beads, from which non-2S rRNA is removed off as supernatant (Citation9). Both methods have proven effective in eliminating 2S rRNA reads, but each requires additional steps and reagents which, in the case of magnetic beads, can be costly. We therefore wanted to determine if depletion could be achieved during 5′ ligation and cDNA synthesis by specifically blocking 2S RNA products from jointly participating in these two reactions. For 2S RNA, this is expected to yield truncated cDNA products lacking sequences complementary to the 5′ adapter. Without these sequences, library PCR amplification is impossible. Blocking with terminator oligos may thus serve as a simple means to eliminate 2S rRNA from the sequencing reaction without additional enzymatic steps or use of magnetic beads.
To test this, we modified a previously published protocol (Citation10) with the intent of precluding 5′ adapter ligation to 2S rRNA. We first size selected for 19 to 30 nt RNAs with 15% acrylamide/urea gels, followed by 3′ adapter ligation. Prior to 5′ adapter ligation, we pre-annealed a 2S rRNA blocker complementary to the 2S rRNA sequence and ending with a 3′ C3 spacer (). Subsequently, we annealed the reverse transcription (RT) primer complementary to the 3′ adapter. This RT primer was annealed prior to 5′ ligation to eliminate adapter-adapter ligation products (Citation10). After these pre-annealing steps, 5′ ligation and RT were done, followed by PCR-based barcode incorporation and Illumina sequencing (see Supplementary Material). For these experiments, two separate size selected small RNA pools (experiment 1 and experiment 2) were extracted from Drosophila virilis ovaries. After gel purification and size selection, the two RNA pools were each split into no block and block treatments.
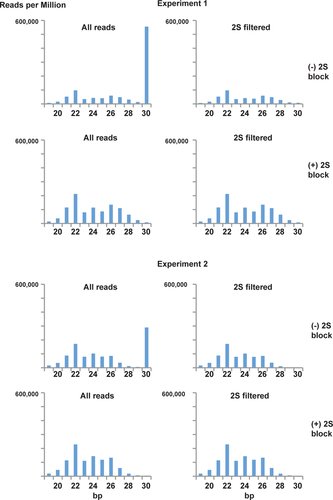
To measure depletion of 2S rRNA, we quantified the proportion of 19 to 30 bp reads mapping to 2S rRNA. indicates that 2S rRNA was nearly entirely eliminated when the 2S block was utilized. Since the number of unique small RNA reads is greater for the blocked samples in both experiments, we can conclude that the block is not leading to PCR compression of library complexity. Examining the size distribution of reads (), one can see that the 2S block reaction largely eliminates the 30 bp class of RNA without perturbing the underlying size distribution. In all cases, a 22 nt peak of miRNA is evident against a background of presumptive piRNA and siRNA.
Table 1. 2S mapped reads from 2S block (-) and (+) small RNA sequence runs.
Percent genome mapping of total reads is in parentheses. The percentage of total reads in 2S reads is also in parentheses.
2S filtered distributions were obtained after removal of 2S mapping sequences.
To determine whether non-specific 2S block hybridization perturbed signatures of miRNA expression from ovaries, we mapped non-2S rRNA small RNA reads against the annotated miRNA library from D. virilis. Comparison of profiles was achieved by measuring the proportion of non-2S reads that mapped to these miRNAs. miRNA profiles were very similar between blocked and non-blocked libraries (,). In experiment 1, there was some difference in the expression profile between two of the most highly expressed miRNAs. However, this was not the case in experiment 2. To directly compare the miRNA profiles between experiments, we used Pearson's correlation coefficient r as a metric (). Among all pairwise comparisons and considering all miRNAs, the pair with the most similar results were Experiment 2 (no 2S block) and Experiment 2 (2S block) (r = 0.999). Overall, results indicate that Experiment 1 (no 2S block) is an outlier since its Pearson's r value is lower with all other samples. However, its miRNA profile is most similar to its own block treatment pair (r = 0.965). Since block pairs and no block pairs were never most similar to each other, we can conclude that blocking treatment did not result in a systematic bias in miRNA profile signatures. Excluding the four highly expressed RNAs, the most similar two pairwise results were achieved within RNA extraction rather than within block/no block treatment.
A) log10-log10 miRNA profiles from two experiments, comparing no block and 2S block treatments, after 2S rRNA sequence filtering. Measurements are in reads per million mapped (RPMM) where mapped (M) is measured from the total number of reads mapping to the D. virilis genome. B) log10-log10 transposable elements (TE) mapping small RNA profiles from two experiments, comparing non block and 2S block treatments. Measurements are the RPMM of non-2S RNA/non-miRNA mapping reads that map to each of the annotated D. virilis TEs. C) miRNA profile similarity comparison as measured by Pearson's r. Values in parentheses are with highly expressed miRNAs excluded. Red indicates most similar pair among a set of comparisons. D) TE small RNA profile similarity. All results, block or no block, are highly similar.
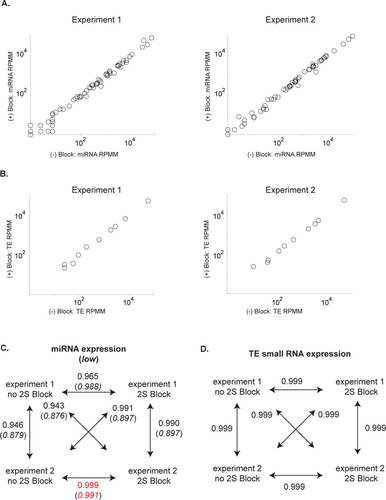
We also determined whether 2S blocking perturbed the signature of small RNAs targeting transposable elements (TEs). We mapped non-2S/non-miRNA reads against the 10 well annotated TEs from D. virilis. The vast majority of those mapping TEs were 23—30 nt in length, indicating that they were piRNAs. TE mapping small RNA profiles are remarkably robust across all treatments and there is no evidence 2S blocking perturbs these profiles (). Interestingly, small RNA profiles from TEs are more robust to experimental variation than miRNA profiles (). This might be explained by the fact that each TE represents many different piRNAs. In contrast to single miRNA measurements, pools of diverse piRNAs may be robust to stochastic variation in cloning, PCR and sequencing efficiency.
To conclude, we have demonstrated a simple and inexpensive method to limit rRNA reads from small RNA sequence reactions. This is most likely achieved by limiting 5′ adapter ligation to the double-stranded rRNA/oligonucleotide duplex. Previous studies have shown that 5′ adapter/3′ adapter ligation events are minimized in the presence of duplex DNA (Citation10). The presence of a 3′ C3 spacer may further contribute to ligation blocking. Since rRNA excess is a general problem for RNA-seq, similar blocking strategies that limit adapter ligation and full length cDNA synthesis may prove generally useful, limiting the need for magnetic beads and additional enzymatic steps.
Author contributions
MLW and JPB designed the experiments. MLW conducted the experiments. JPB wrote the paper.
Competing interests
The authors declare no competing interests.
RNA Isolation and library preparation
Download PDF (245.6 KB)Acknowledgments
Funding for this work was provided by NSF Award MCB-1022165 and NIH P20GM103638. This paper is subject to the NIH Public Access Policy
Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at: www.tandfonline.com/doi/suppl/10.2144/000114102
Additional information
Funding
References
- Adiconis, X., D.Borges-Rivera, R.Satija, D.S.Deluca, M.A.Busby, A.M.Berlin, A.Sivachenko, D.A.Thompson, et al.. 2013. Comparative analysis of RNA sequencing methods for degraded or low-input samples. Nat. Methods10:623–629.
- Morlan, J.D., K.Qu, and D.V.Sinicropi. 2012. Selective depletion of rRNA enables whole transcriptome profiling of archival fixed tissue. PLoS ONE7:e42882.
- Huang, R., M.Jaritz, P.Guenzl, I.Vlatkovic, A.Sommer, I.M.Tamir, H.Marks, T.Klampfl, et al.. 2011. An RNA-Seq strategy to detect the complete coding and non-coding transcriptome including full-length imprinted macro ncRNAs. PLoS ONE6:e27288.
- Yi, H., Y.J.Cho, S.Won, J.E.Lee, H.Jin Yu, S.Kim, G.P.Schroth, S.Luo, and J.Chun. 2011. Duplex-specific nuclease efficiently removes rRNA for prokaryotic RNA-seq. Nucleic Acids Res.39:e140.
- Ramsköld, D., S.Luo, Y.C.Wang, R.Li, Q.Deng, O.R.Faridani, G.A.Daniels, I.Khrebtukova, et al.. 2012. Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat. Biotechnol.30:777–782.
- Tariq, M.A., H.J.Kim, O.Jejelowo, and N.Pourmand. 2011. Whole-transcriptome RNAseq analysis from minute amount of total RNA. Nucleic Acids Res.39:e120.
- Zhuang, F., R.T.Fuchs, and G.B.Robb. 2012. Small RNA expression profiling by high-throughput sequencing: implications of enzymatic manipulation. J. Nucleic Acids2012:360358.
- Li, C., V.V.Vagin, S.H.Lee, J.Xu, S.M.Ma, H.L.Xi, H.Seitz, M.D.Horwich, et al.. 2009. Collapse of Germline piRNAs in the Absence of Argonaute3 Reveals Somatic piRNAs in Flies. Cell137:509–521.
- Seitz, H., M.Ghildiyal, and P.D.Zamore. 2008. Argonaute loading improves the 5′ precision of both MicroRNAs and their miRNA* strands in flies. Curr. Biol.18:147–151.
- Vigneault, F., D.Ter-Ovanesyan, S.Alon, S.Eminaga, D.C.Christodoulou, J.G.Seidman, E.Eisenberg, and G.M.Church. 2012. High-throughput multiplex sequencing of miRNA. Curr Protoc Hum Genet.Chapter 11:11–10 Unit 11 12.

