
A common operation in molecular biology involves the ligation or “stitching” of two or more adjacent sequences of DNA. In efforts directed towards the synthesis of a large number of very long polyketide synthase genes, we required a process to efficiently stitch multiple synthetic DNA fragments of 500–800 bp together to produce segments of up to 6 kbp. Multiple ligations of fragments are often performed one at a time into a vector, requiring n − 1 (where n is the number of fragments) sequential cycles of cleavage, purification, and cloning for each of the inserts. Furthermore, because the fragment is produced or excised employing different restriction sites, the vector into which it is cloned must likewise contain the same restriction sites; this often requires specialized linkers that contain multiple cloning sites (MCS) co-linear with the fragments to be inserted. Efficiencies may be gained by parallel processing the ligation cycles, but significant time and effort is invested in isolation, purification, and cloning of inserts, which is scaled upwards by the number of large DNA sequences being prepared simultaneously.
In the present work, we describe a method, termed ligation by selection (LBS), for ligating multiple adjacent DNA fragments that does not require intermediate fragment isolation or specialized MCS linkers and is amenable to parallel processing and semi-automation. Fragments that ultimately are to lie adjacent to each other are each cloned into separate plasmid vectors that have a common antibiotic marker, a linking restriction site for joining the fragments, and a remote restriction site on the vector; in addition, each has a unique site to be used for restriction-purification (Citation1) and a unique antibiotic marker (). Each vector is cleaved at the common restriction site and the remote site common to both. One of the four resultant fragments is further destroyed at its unique restriction-purification site. The crude digestion products are ligated in situ to generate a single plasmid containing the two fragments that can be selected for directly. Two of the two-fragment plasmids are then ligated to make a plasmid with a four-fragment insert, and the process is continued recursively to lengthen the size of the insert in each cycle. Although the method has been developed to stitch together synthetic genes, a modified version could as well be used to ligate multiple DNA fragments from other sources (e.g., PCR).
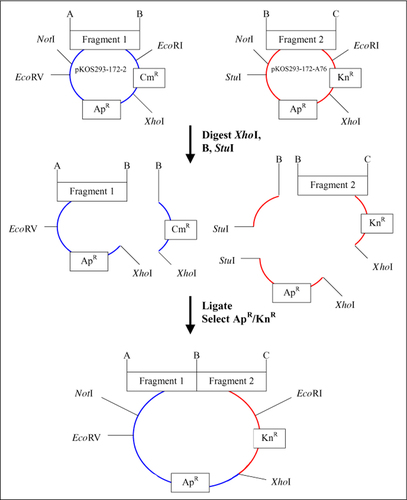
Modified pUC18 sister cloning vectors pKos293-172-2 (3.6 kb; insert acceptor) and pKos293-172-A76 (3.9 kb; insert donor), showing restriction sites at the insert edges (A, B, and C). Both vectors contain a common XhoI site used for assembly of inserts. Vector pKos293-172-2 contains a unique CmR marker and EcoRV restriction site, while vector pKos293-172-A76 contains a unique KnR marker and StuI restriction site. For LBS, both acceptor and donor vectors are digested at the XhoI site and B site, the common site between the two adjacent fragments. The donor vector, pKos293-172-A76 in this example, is treated with StuI. The desired two fragments are ligated together, selected on Ap and Kn, and analyzed by gel electrophoresis after restriction with NotI/EcoRI. LBS, ligation by selection; CmR, chloramphenicol resistance gene; KnR, kayamycin resistance gene; ApR, ampicillin resistance gene.
All enzymes were obtained from New England Biolabs (Beverly, MA, USA) and used as recommended. Molecular biological techniques were performed using standard protocols (Citation2). Synthetic DNA fragments were designed and prepared by a modification of reported methods (Citation3–5).
A linker duplex for uracil-DNA glycosylase-based ligation-independent cloning (UDG-LIC) (Citation6,Citation7) comprised of oligonucleotides UDG-L1 and UDG-L2, which contained a 5′ overhang [nucleotides (nt) 1–4] compatible with HindIII and a 3′ EcoRI overhang (nt 66–69), was introduced into HindIII/EcoRI-digested pUC18 to give pKOS293-118-72. The linker contained a SacI site (nt 36–41), a NotI site (nt 6–13), and two N. BbvCIA sites (nt 14–20 and 58–64), and upon introduction destroyed the HindIII site. The chloramphenicol resistance gene (CmR) was amplified from pACYC184 with the primers Cm-forward and Cm-reverse (). The kanamycin resistance gene (KnR) was amplified from pET35b with primers Kan-forward and Kan-reverse. The PCR fragments containing CmR and KnR were each blunt-ligated into pKOS293-118-72, which was linearized with SapI and treated with Klenow fragment of DNA polymerase I. Orientations were determined by fragment analysis of XhoI/NotI digests, and plasmids containing the XhoI site between the unique marker and the ampicillin resistance gene (ApR) marker were named pKOS293-135-52 (CmR) and pKOS293-149-18 (KnR).
Table 1. Oligonucleotide Primers and Linkers
Next, a linker duplex using oligonucleotides EcoRV L-1 and EcoRV L-2 containing an EcoRV site (nt 15–20), a 5′ overhang (nt 1–2) compatible with NdeI, and a 3′ overhang (nt 35–38) compatible with KasI was introduced into the NdeI/KasI-digested CmR vector (pKOS293-135-52) to give pKOS293-172-2, with destruction of NdeI/KasI cloning sites. An analogous duplex using oligonucleotide linkers StuI L-1 and StuI L-2 containing the StuI site at nt 15-20 was inserted into the NdeI/KasI-digested KnR vector (pKOS293-149-18) to give pKOS293-172-A76.
The digest solution contained 6 µL of the acceptor plasmid (100–200 ng), 3 µL of bovine serum albumin (BSA) (250 µg/mL), 1 µL (20 U) of XhoI, 3 µL of 10× New England Biolabs buffer appropriate for the restriction enzyme used, 1 µL of the restriction enzyme to cleave the 3′ edge of the acceptor fragment and 5′ edge of the donor fragment, and water to give a final volume of 30 µL. The donor plasmid was digested in the same manner, except 20 U EcoRV or 10 U StuI were added, depending on which restriction site was present. Samples were digested at 37°C for 2 h, heated for 20 min at 80°C, and analyzed by gel electrophoresis to verify digestion of the parent plasmids. Restriction enzymes used successfully to cut the 5′ and 3′ fragment edges included AgeI (5 U/µL), AvrII (4 U/µL), BglII (10 U/µL), BsiWI (10 U/µL), BssHII (4 U/µL), Kpn I (10 U/µL), NgoMIV(10 U/µL), NheI (10 U/µL), PstI (20 U/µL), SacII (20 U/µL), and SphI (5 U/µL).
The ligation solution contained 3 to 4 µL (10–30 ng) of each of the digested donor and acceptor plasmids, 1.5 µL (600 U) of T4 ligase, and water to 30 µL. After 2 h at room temperature, 2 µL of the ligation solution was used to transform DH5α, and the mixture was streaked onto a plate containing carbenicillin and either kanamycin or chloramphenicol (depending on the donor vector used). Plasmids isolated from clones were digested with 5 U NotI/10 U Eco-RI for 2 h at 37°C and analyzed by gel electrophoresis. Plasmids containing the inserts of appropriate size were purified and used for the next LBS cycle.
pUC18 was modified to give the two sister vectors, pKOS293-172-2 and pKOS293-172-A76 (). The original MCS was replaced with a sequence to facilitate UDG-LIC cloning of inserts, but any approach that permits cloning of PCR fragments would suffice. For example, with our vectors, the NotI and EcoRI sites could be used to accept PCR fragments generated by introducing these sites on the primer ends. The two vectors differ in that each contain a unique restriction site (EcoRV or StuI) 5′ of the cloning site, and a unique antibiotic resistance marker (CmR or KnR) 3′ from the cloning site. Both vectors contain the ApR of pUC18, a 5′ NotI site and 3′ EcoRI site flanking the cloning site, and an XhoI site between the ApR and unique antibiotic markers.
The general approach is first described for a single LBS cycle for the joining of two DNA fragments cloned into the two sister plasmids. The plasmid containing the 5′ fragment is referred to as the acceptor and that containing the 3′ fragment is referred to as the donor. The common restriction site used to join the fragments exists at the 3′ end of the acceptor fragment and the 5′ end of the donor fragment. As shown in , two sister vectors containing adjacent inserts are excised at the XhoI site and at the linking restriction site. The acceptor vector is thus cleaved to a large fragment containing the insert and a small fragment containing the unique marker. The donor vector is cleaved to a small fragment containing the insert and the unique marker and a large fragment containing a unique restriction site (StuI or EcoRV), which is further cleaved to provide restriction purification and prevent subsequent ligation of this fragment. The mixture of fragments is then treated with T4 ligase and transformed into Escherichia coli, and colonies are selected for resistance to the unique marker derived from the donor fragment and the common ApR marker. The only impurity expected is the single-fragment donor plasmid that survived restriction purification and religated. Analysis of the transformants is performed by cleaving the product with NotI/EcoRI, followed by gel electrophoresis. The insert derived from the product is the sum of the sizes of the original inserts and is easy to discern from smaller surviving donor vector. One LBS cycle requires about 3 days. On day one, the cleavage and ligation reactions are performed, and the bacteria are transformed and plated. Day 2 involves colony picking and cell growth, and day 3 involves plasmid preparation and insert size analysis.
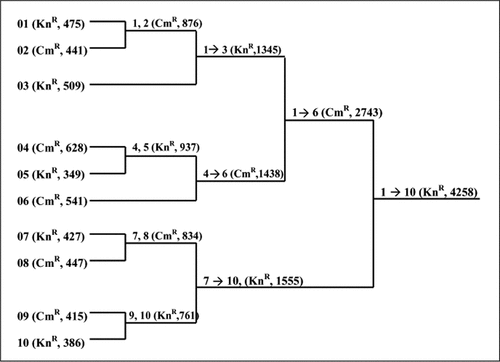
To utilize LBS recursively, a dendrographic plan is prepared that accounts for the alternation of resistance markers and restriction-purification sites of acceptor and donor vectors that occur in each LBS cycle, and minimizes the number of cycles required in parallel processing. shows the plan used for connecting ten DNA fragments together to form a 4.3-kbp module of a polyketide synthase gene. As shown, the experiment requires nine ligations that can be parallel processed in four cycles. First, ten single DNA fragments of 349–628 bp were cloned into the appropriate sister plasmid. In the first cycle, eight of these were connected to form four two-fragment plasmids. In the next cycle, the two remaining single-fragment inserts were ligated to two of the two-fragment inserts, and two of the two-fragment inserts were ligated to give four-fragment inserts. In cycle 3, the 1345- and 1438-bp inserts were ligated to give the 2743-bp fragment, which was then ligated to a 1555-bp fragment to give the final 4258-bp gene. Eight colonies from each of the ligation reactions were analyzed to determine the success rate of ligations. Five of the ligations had 8/8 correct inserts, three had 7/8 and one had 1/8. Overall, 83% of the plasmids examined had the appropriate insert, and the failures were all unchanged donor plasmid. We have used this approach to perform 102 ligations of adjacent DNA fragments, making DNA sequences greater than 6.5 kbp. Of clones examined, approximately 75% were the expected ligated products, while the remaining 25% were donor vector that contained the marker and survived restriction purification. Of 12 restriction enzymes used to cleave fragment ends, 11 were validated as acceptable for this method; only one, AflII, was rejected, in that its use required fragment purification for successful ligation.
In parentheses are shown the unique resistance marker used for selection and the insert size for each LBS cycle. LBS, ligation by selection.
Major advantages of LBS over conventional ligation of inserts are: (i) avoidance of the need to isolate, purify, and ligate individual fragments; (ii) evasion of the need for specialized MCS linkers; and (iii) the ease with which parallel processing of operations may be applied. The example provided of ligating ten sequential fragments would have required design and construction of a MCS linker and some nine fragment isolation, purification, and cloning steps. While these advantages may not be significant in the preparation of a single long DNA sequence, they represent a major consumption of time and effort upon scale-up to many long DNA sequences. A shortcoming of LBS involves the use of 6-bp restriction sites on the plasmid for cleavage (XhoI) or restriction purification (StuI or EcoRV), since their presence in the inserts must be avoided. In the next generation of LBS vectors, we will circumvent this potential problem by using enzymes with longer recognition sequences, such as the recently available homing endonucleases.
Acknowledgments
This work was supported in part by National Institute of Standards and Technology Advanced Technology Program Grant Award No. 70NANB2H3014. The authors would like to thank Ralph Reid, Sebastian Jayaraj, and Kedar Patel for assistance and advice.
References
- Wells, J.A., B.C.Cunningham, T.P.Graycar, and D.A.Estell. 1986. Importance of hydrogen bond formation in stabilizing the transition state of subtilisin. Phil. Trans. R. Soc. Lond. A317:415–423.
- Sambrook, J., E.F.Fritsch, and T.Maniatis. 1989. Molecular Cloning: A Laboratory Manual, 2nd ed.CSH Laboratory Press, Cold Spring Harbor, NY.
- Prodromou, C. and L.H.Pearl. 1992. Recursive PCR: a novel technique for total gene synthesis. Protein Eng.5:837–829.
- Stemmer, W.P.C., A.Crameri, K.D.Ha, T.M.Brennan, and H.L.Heyneker. 1995. Single-step assembly of a gene and entire plasmid from large numbers of oligodeoxyribonucleotides. Gene164:49–53.
- Hoover, D.M. and J.Lubkowski. 2002. DNAWorks: an automated method for designing oligonucleotides for PCR-based gene synthesis. Nucleic Acids Res.30:e43.
- Nisson, E.N., A.Rashtchian, and P.C.Watkins. 1991. Rapid and efficient cloning of alu-PCR products using uracil DNA glycosylase. PCR Methods Appl.2:120–123.
- Rashtchian, A., G.W.Buchman, D.M.Schuster, and M.S.Berninger. 1992. Uracil DNA glycosylase-mediated cloning of polymerase chain reaction-amplified DNA: application to genomic and cDNA cloning. Anal. Biochem.206:91–97.

