
It is often necessary to conduct genetic or genomic analysis with a limiting amount of DNA. Whole genome amplification based on the multiple displacement capacity of DNA polymerases such as φ29 DNA polymerase (multiple displacement amplification; MDA) has been successfully used to amplify eukaryotic genomic DNA with minimum bias (Citation1–3). Although the use of MDA has been limited so far to the genomic analysis of eukaryotic organisms, there may be useful applications of this method to prokaryotic genomes. For bacterial species that can be cultivated in laboratory conditions, pure genomic DNA of a single strain can be feasibly obtained in sufficient amounts necessary for genomic analyses such as genome DNA library construction, molecular typing or genome sequencing. However, more than 99% of microbial species in nature are refractory to cultivation in laboratory growth conditions (Citation4). Consequently, knowledge of the genomic contents of microorganisms has been largely biased toward those that are culturable. In an effort to increase our understanding of the unculturable microorganisms, several strategies have been described (Citation4). MDA is a promising tool to obtain a sufficient amount of genomic DNA from unculturable microbial cells in the environment. For example, the genome sequencing of the unculturable bacterium Epulopiscium is currently underway and being made possible in part by the MDA reaction to obtain genomic DNA (Citation5).
For most genomic analyses of unculturable species, it would be necessary to obtain pure genomic DNA from a single strain without contaminating DNA. Such genomic DNA might be obtained by MDA from a single cell or a cluster of a single strain in an environmental niche. For such purposes, using bacterial cells rather than purified genomic DNA as templates in the MDA reaction would be beneficial to maximize the yield of DNA during purification steps. For PCR amplification, template DNA is usually obtained by heating bacterial cells (e.g., 95°C for 2–5 min) to release genomic DNA as well as plasmid DNA. However, it was shown that the heating step could cause depurination of the DNA molecules making them unsuitable as templates for PCR amplification (Citation6). The heating of genomic DNA at 95°C also resulted in a significant decrease in the yield of MDA products in an eukaryotic system (Citation1). In this study, denaturation by heating also resulted in lower locus representation, which is defined as the locus copy number in 1 µg of amplified DNA divided by the locus copy number in 1 µg of genomic DNA control. With denaturation by heating, the locus representation ranged from 2%–80%, while 80%–225% was obtained with nondenatured genomic DNA (Citation1). It was previously demonstrated that the presence of PCR buffer during the heating step prevented depurination in DNA templates, which resulted in better efficacy in PCR amplification (Citation6). In this study, we found that heating bacterial cells in the presence of PCR buffer resulted in DNA templates that increased the efficacy of MDA reactions.
An overnight culture of Salmonella enteritidis LK5 (Citation7) grown in Luria bertani (LB) broth was serially diluted in sterile water. Viable cells were counted by plating the serial dilutions on LB agar plates. The dilutions were used as templates in MDA reactions using the whole genome amplification kit REPLI-g 625S (Molecular Staging, New Haven, CT, USA). A 2-µL aliquot of each dilution containing approximately 600, 60, and 6 colony-forming units (cfus) was mixed with either 0.5 µL double-distilled water or 0.5 µL 10× cloned Pfu DNA polymerase buffer [200 mM Tris-HCl, pH 8.8, 20 mM MgSO4, 100 mM KCl, 100 mM (NH4)2SO4, 1% Triton® X-100, 1 mg/mL nuclease-free bovine serum albumin (BSA)], which was obtained from Stratagene (La Jolla, CA, USA), to 2× final concentration. The cell suspensions were heated for 10 min at 98°C to release genomic DNA and cooled to 4°C, at which time 47.5 µL of a master mixture containing 34.5 µL of double-distilled water, 12.5 µL of 4× master mixture, and 0.5 µL of φ29 DNA polymerase were added to each tube. Then the reactions were incubated at 30°C for 16 h, followed by an incubation at 65°C for 3 min to inactivate the φ29 DNA polymerase. The reactions were then stored at 4°C and analyzed by gel electrophoresis using 0.6% agarose gel.
shows the whole genomic DNA amplified from bacterial cells after heating in the presence of double-distilled water. Whole genomic DNA was amplified from both MDA reactions containing 600 and 60 cfus (, lanes 2 and 3, respectively), but the yield of the MDA product was lower with 60 cfus. In order to determine the quality of the amplified DNA, we used the MDA products as templates for PCR amplification of a genetic locus that is specific to S. enteritidis (Citation8). The PCR mixture contained 1.25 µL dimethylsulfoxide (DMSO), 1.25 µL 25 mM dNTPs, 1.0 µL of each sdf I-F and sdf I-R primers (350 ng/µL; see ), and 0.2 µL Taq DNA polymerase (Applied Biosystems, Foster City, CA, USA) in a 25-µL reaction. The reactions were heated at 95°C for 2 min and amplified through 30 cycles of 95°C for 30 s, 58°C for 1 min, and 70°C for 1 min. After a final extension at 70°C for 10 min, the reactions were kept at 4°C. As shown in , PCR products of 294 bp were amplified from both MDA products obtained from 600 and 60 cfus (, lanes 4 and 5, respectively), while the efficiency was much lower with 60 cfus.
Whole genomic DNA was amplified from approximately 600 (lane 2) and 60 (lane 3) colony-forming units (cfus) after releasing genomic DNA by heating at 98°C for 10 min. S. enteritidis-specific fragments (294 bp) were amplified from the respective MDA products (lanes 4 and 5). Purified S. enteritidis LK5 genomic DNA was used as positive control to amplify sdf I fragment indicated by the arrow (lane 6). A 1-kb DNA ladder (Invitrogen, Carlsbad, CA, USA) was used as a standard marker (M).
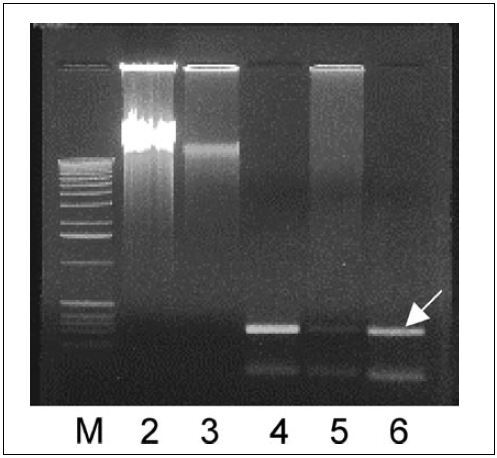
Table 1. PCR Primers and Sequences
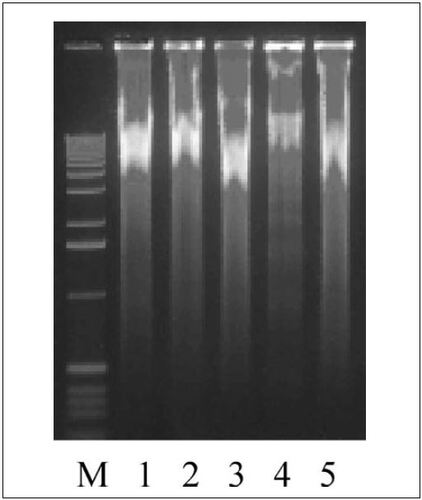
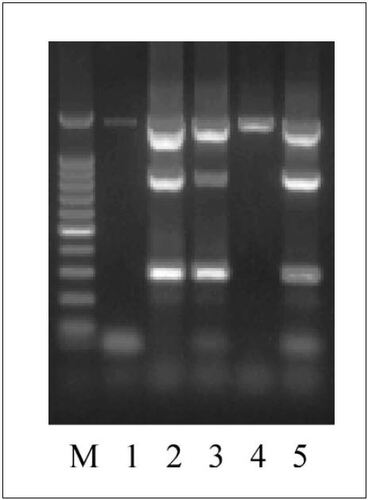
In order to determine whether the presence of PCR buffer could improve the efficacy in MDA reactions, the MDA reactions were performed as before, using 2 µL of serial dilutions containing approximately 600, 60, and 6 cfus, except that dilutions were heated for 98°C for 10 min in the presence of 2× cloned Pfu DNA polymerase buffer. There was no gross difference in the yield of MDA products amplified by heating with and without 2× PCR buffer (). It was previously reported that MDA reactions initiated with very small inputs of genomic DNA tend to be contaminated with spurious DNA sequences obtained by primer-directed DNA synthesis (Citation2). To determine whether the presence of the 2× PCR buffer during the heating step had effects on the quality of the MDA products, three unlinked genetic loci were simultaneously amplified in multiplex PCRs as described above using those MDA products as templates. The three sets of primers used in the multiplex PCRs () were: (i) sdf I-F and sdf I-R primers for sdf I fragment (Citation8); (ii) 63F and 1389R for 16S rRNA gene (Citation9); and (iii) rpoS-F and rpoS-R for the promoter region and 5′ coding sequence of rpoS gene. As shown in , the presence of 2× PCR buffer during heating to release genomic DNA greatly improved the quality of the resulting MDA products. From the MDA product obtained from 600 cfus in the absence of 2× PCR buffer (, lane 1), only the 16S rRNA gene sequence was amplified. In contrast, the MDA products obtained from 600 and 60 cfus in the presence of 2× PCR buffer allowed amplification of all three fragments (, lanes 2 and 3). The 16S rRNA gene fragment was amplified even from the MDA product obtained from 6 cfus in the presence of 2× PCR buffer (, lane 4).
Whole genomic DNA was amplified from approximately 600 (lanes 1 and 2), 60 (lane 3), and 6 (lane 4) colony-forming units (cfus). All samples were heated at 98°C for 10 min in the presence of 2× PCR buffer before the MDA reactions except lane 1, which contained double-distilled water instead. Whole genomic DNA was also amplified using the genomic DNA purified from Salmonella enteritidis LK5 as a template (lane 5). A 1-kb DNA ladder (Invitrogen) was used as a standard marker (M).
MDA products amplified from approximately 600 (lanes 1 and 2), 60 (lane 3), and 6 (lane 4) colony-forming units (cfus) were used as templates to amplify three loci in multiplex PCR (294 bp for sdf I; 800 bp for rpoS promoter; 1300 bp for 16S rRNA gene). The MDA reactions were performed using genomic DNA released by heating at 98°C for 10 min in the presence of 2× PCR buffer except lane 1, which contained double-distilled water instead. S. enteritidis LK5 genomic DNA was used as a positive control for the mutiplex PCR (lane 5). A 100-bp DNA ladder (Promega, Madison, WI, USA) was used as a standard marker (M).
In summary, the results of this report suggests that the addition of PCR buffer to 2× final concentration during the heating step to release genomic DNA from bacterial cells significantly improved the quality of the MDA products. We expect that this improvement in the MDA reaction will help minimize the number of bacterial cells needed to amplify whole genomic DNA. The availability of genomic DNA of unculturable bacterial species by the improved MDA protocol will facilitate characterization of their genomic contents. The result also suggests that it may be possible to amplify whole genomic DNA with minimum bias even from a single bacterial cell after appropriate improvements are made in the MDA protocol.
References
- Dean, F.B., S.Hosono, L.Fang, X.Wu, A.F.Faruqi, P.Bray-Ward, Z.Sun, Q.Zong, et al.. 2002. Comprehensive human genome amplification using multiple displacement amplification. Proc. Natl. Acad. Sci. USA99:5261–5266.
- Lage, J.M., J.H.Leamon, T.Pejovic, S.Hamann, M.Lacey, D.Dillon, R.Segraves, B.Vossbrinck, et al.. 2003. Whole genome analysis of genetic alterations in small DNA samples using hyperbranbched strand displacement amplification and array-CGH. Genome Res.13:294–307.
- Gorrochotegui-Escalante, N., W.C.BlackIV. 2003. Amplifying whole insect genomes with multiple displacement amplification. Insect Mol. Biol.12:195–200.
- Torsvik, V. and L.Øvreås. 2002. Microbial diversity and function in soil: from genes to ecosystems. Curr. Opin. Microbiol.5:240–245.
- Nelson, K.E. 2003. The future of microbial genomics. Environ. Microbiol.5:1223–1225.
- Frost, M.R. and J.A.Guggenheim. 1999. Prevention of depurination during elution facilitates the reamplification of DNA from differential display gels. Nucleic Acids Res.27:e6.
- Edwards, R.A, D.M.Schifferli, and S.R.Maloy. 2000. A role for Salmonella fimbriae in intraperitoneal infections. Proc. Natl. Acad. Sci. USA97:1258–1262.
- Agron, P.G., R.L.Walker, H.Kinde, S.J.Sawyer, D.C.Hayes, J.Wollard, and G.L.Andersen. 2001. Identification by subtractive hybridization of sequences specific for Salmonella enterica serovar Enteritidis. Appl. Environ. Microbiol.67:4984–4991.
- Osborn, A.M., E.R.Moore, and K.N.Timmis. 2000. An evaluation of terminal-restriction fragment length polymorphism (T-RFLP) analysis for the study of microbial community structure and dynamics. Environ. Microbiol.2:39–50.

