
Abstract
Well-characterized epidemiological resources are generated with great effort, yet associated patient DNA samples can be limiting. The efficacy of the whole genome amplification (WGA) method, termed multiple displacement amplification (MDA), was assessed for detecting heterozygous sequence variants, mutation scanning, and PCR for challenging segments. Fifteen common polymorphisms from 10 genes located on 8 chromosomes were genotyped by direct sequencing of 300 PCR products from 115 high-quality MDA-amplified DNA samples extracted by different methods. The GC content of these analyzed segments ranges from 30% to 69%. Genotyping results demonstrate 100% accuracy. For heterozygotes, the relative intensity of peaks generated by the two alleles is highly similar for genomic and MDA-amplified genomic DNA, independent of GC content. In contrast, one of four heterozygous loci was mistyped when lower quality MDA-amplified DNA samples were used. The results of single-stranded conformation polymorphism (SSCP)-type of mutation scanning for seven MDA-amplified DNA samples in four genes were concordant with the genomic DNA samples. PCR on MDA-amplified DNA was routinely successful for challenging 10- and 12-kb segments with GC content ranging from 30% to 80%, demonstrating that rather long segments, which are difficult to amplify with PCR, are amplified well with MDA. These results suggest that MDA is an effective method of WGA with utility in molecular epidemiology. Quality control of the MDA-amplified DNA is critical for high performance.
Introduction
Human genomic DNA can be limiting in molecular epidemiological and diagnostic analyses. Thus, whole genome amplification (WGA) can be advantageous if the method is comprehensive and robust and if the frequency of false negatives is low.
Existing PCR-based WGA methods, like degenerate oligonucleotide-primed PCR (DOP-PCR) or primer extension preamplification (PEP), suffer from nonspecific amplification artifacts (Citation1), incomplete coverage of loci (Citation2), and DNA <1 kb long (Citation1,Citation3,Citation4). DOP-PCR specifically suffers from substantial bias between loci (Citation5). Furthermore, the PCR products will act as preferential templates, and hence, contamination with WGA-amplified DNA is a risk. PEP offers a relatively low level of amplification, estimated at 100- to 200-fold.
Recently, a robust WGA method based on multiple displacement amplification (MDA) was developed to amplify genomic DNA in an isothermal reaction. When MDA is performed with φ29 DNA polymerase and random exonuclease-resistant primers, 10,000-fold amplification yielding high-fidelity DNA can be achieved (Citation5). Dean et al. (Citation5) applied MDA to human genomic DNA, yielding an average DNA product >10 kb in length with relatively uniform amplification across the genome.
Does MDA come close to achieving the alchemist's dream of converting small amounts of a precious product to large amounts of that product? Much work may be required to address this question, as the 3 billion nucleotides and many sequence motifs of the human genome are analyzed in multiple ways.
Initial studies suggest that MDA is a promising method (Citation5–7). Genotyping by TaqMan® real-time PCR assays has a 99.95% accuracy. Out of a total of 3814 genotype calls compared between nonamplified genomic DNA and MDA-amplified DNA, two discrepancies (0.05%) were found (Citation7). DNA sequencing and reanalysis with TaqMan suggest that the discrepancies were most likely caused by genotyping error and not the result of bias introduced during the MDA process (Citation7). These indicate that MDA-amplified DNA was an accurate representation of genomic DNA, at least for the six single nucleotide polymorphisms (SNPs) assayed.
For molecular epidemiological analyses, some bias in interlocus amplification can be tolerated, provided sufficient DNA is used so that a good signal is seen robustly in the genotyping assay. The relative amplification of the paternal and maternal alleles is key.
Work to date has focused on relative amplification of segments and genotyping known as SNPs with TaqMan assays (Citation5,Citation6). However, in candidate gene mutation analysis, DNA sequencing or mutation scanning methods are used to search for unknown sequence variants. The utility of MDA-amplified DNA for candidate gene mutation analysis has not been assessed.
To assess the utility of MDA in molecular epidemiology with emphasis on candidate gene analyses for predisposition to complex disease, we addressed four key issues: (i) faithful representation of heterozygous genomic variants by DNA sequence analysis with minimal amplification bias; (ii) the effect of GC content; (iii) the ability to detect rearrangements requiring PCR amplification of 10- to12-kb fragments, in which GC content varies from 30% to 80%; and (iv) the actual performance of MDA-amplified DNA in a mutation scanning project.
Materials and methods
DNA Extraction Methods
DNA was extracted either with phenol/chloroform according to the protocol described in Gustafson et al. (Citation8) or with ATGC (Analytical Genetic Testing Center, Denver, CO, USA) or Puregene® (Gentra Systems, Minneapolis, MN, USA) kits according to the instructions of the manufacturer.
Multiple Displacement Amplification
Using multiple displacement amplification, WGA was performed by the Molecular Staging (New Haven, CT, USA) service. WGA was performed in 750 µL using 18 µL of input DNA. A total of 252 genomic DNA samples extracted by three different methods were amplified by multiple displacement amplification. DNA samples were quantitated using the PicoGreen™ assay (Molecular Probes, Eugene, OR, USA) before and after MDA (Citation9,Citation10). Amplification product quality was tested using two TaqMan loci by the Molecular Staging service. Molecular Staging performs a quality assessment (QA) by performing TaqMan analysis of the amplified DNA and by applying an empirically developed algorithm that can predict the genotyping performance of the amplified DNA. One hundred fifteen samples with high-quality (highly usable) amplification and three samples with lower quality (limited utility or unusable) amplification were chosen from the 252 samples for subsequent analyses. These samples had known heterozygous alleles.
Quality Control
In essence, two TaqMan assays for copy number were performed in the MDA-amplified DNA. Based on these two values relative to original genomic DNA standard, MDA-amplified DNA are scored as red (poor quality), yellow (moderate quality), or green (high quality) based on empirical proprietary algorithm. The following details the protocol for TaqMan loci representation assay for Loci 1004 and 699 for MDA-amplified DNA.
PicoGreen quantitation of amplified DNA
A 1/1000 dilution of the amplified genomic DNA was quantitated using PicoGreen double-stranded DNA quantitation reagent (Molecular Probes) as per manufacturers instructions. This value was used to calculate locus representation (see below).
Reagent preparation and TaqMan assay setup
The following reagents were prepared for the TaqMan assay for the QA of 84 MDA products in a single 96-well plate (Molecular Staging patent pending). Platinum Taq DNA polymerase (5 U/µL; Invitrogen, Carlsbad, CA, USA) was thawed on ice. The amplification buffer (26.1 µL/reaction) consisted of 5 µL 10× Taq DNA polymerase PCR buffer (Invitrogen), 5 µL 50 mM MgCl2, 5 µL 10 mM dNTPs, 1 µL 50× ROX (Invitrogen), and 10.1 µL double-distilled water. The 1004 assay mixture contained the WIAF 1004 probe (6.75 µM), the WIAF 1004 forward primer (8.5 µM), and the WIAF 1004 reverse primer (8.5 µM). The 699 assay mixture contained the WIAF 699 probe (6.75 µM), the WIAF 699 forward primer (8.5 µM), and the WIAF 699 reverse primer (8.5 µM). All were prepared and placed on ice.
Oligonucleotides
The WIAF 1004 (chromosome 2) probe sequence is 5′-GATGGCATTACTGGCACTTTGAGTTTTAC-3′, and the primer sequences are forward, 5′-GTCTTTAGCTGCTGAGGAAATG-3′; reverse, 5′-AGCAGAATTCTGCACATGACG-3′.
The WIAF 699 (chromosome 17) probe sequence is 5′-TGAACTGCTCCTTGGCAGGGATTT-3′, and the primer sequences are forward, 5′-TGCTCCCTGTCCCATCTG-3′; reverse, 5′-AGACAGTATGCCTTTATTTCACCC-3′.
Standard curve
A four-point genomic DNA standard curve was prepared by diluting control human genomic DNA (Promega, Madison, WI, USA) via serial dilutions with distilled water to 0.5, 0.05, 0.005, and 0.0005 µg/20 µL. A total volume of 60 µL was prepared for each standard in 1.5-mL Eppendorf® tubes. Duplicates of each 20-µL standard were transferred to a 96-well plate (row H) for a total of eight standards (0.5 µg, wells H1 and H5; 0.05 µg, wells H2 and H6; 0.005 µg, wells H3 and H7; and 0.0005 µg, wells H4 and H8).
Amplified samples
Amplified samples were diluted 1/100, and 20 µL of the diluted product were transferred into the 96-well plate (rows A–G).
Control amplifications
Three control amplifications from 10 ng control DNA (Promega) were included in each plate of amplified DNA. Twenty microliters of each of three 1/100 diluted control MDA reactions were pipeted into the 96-well plate (row H wells 9, 10, and 11).
A QA master mixture was prepared for 100 reactions in a 5-mL round-bottom tube by mixing amplification buffer, locus 1004 primer/probe mixture, locus 699 primer/probe mixture, and platinum Taq DNA polymerase. The QA master mixture was mixed by inverting the tube five times and then stored on ice. Thirty microliters of the QA master mixture was added to each well with experimental MDA product, control MDA product, and standards. The 96-well plate was covered with an optical adhesive cover, centrifuged at 200× g for 1 min, and placed in an ABI Prism® 7900 Sequence Detection System (Applied Biosystems, Foster City, CA, USA). Amplification was performed under the following conditions: 50°C for 2 min, 95°C for 10 min, followed by 40 cycles of 95°C for 15 s, and 60°C for 1 min.
After the 7900 Sequence Detector runs were complete, cycle threshold (Ct) values were obtained for all samples using the ABI software (Applied Biosystems) as per the manufacturer's instructions. Ct is defined as the PCR cycle where the fluorescent signal from the amplification rises above background. One cycle difference in Ct is equal to a 2-fold difference in target copy number.
Quality Assessment Criteria
A standard curve was generated using the Ct from the four-point genomic DNA standards. The x-axis depicted the log[control DNA in µg], and the y-axis displayed the Ct value. The minimum R2 value for the standard curve was 0.9900. If R2 values were lower than 0.9900, the assay was repeated. An equation for the standard curve was generated in the form y = mx + b, where y is the Ct, and x is log in micrograms of input DNA. Locus representation was reported as a percentage and was calculated for both loci as follows: (i) the Ct value obtained for each amplified sample was plugged into the standard curve equation as the “y” value, and a value for the theoretical micrograms of input DNA (antilog “x”) was obtained; and (ii) locus representation for each sample was calculated by taking the ratio of theoretical micrograms input DNA (as calculated using the standard curve/µg input-amplified DNA as measured by PicoGreen) × 100.
A value of 100% indicates that the locus was represented equally in the amplified and nonamplified DNA (Citation6). All samples were sorted into one of the three categories listed below based on locus representation: (i) highly usable, both loci with greater than 3% locus representation with a ratio between representation at both loci of less than 20-fold; (ii) limited utility, one or both loci between 0.1% and 3% representation and/or a ratio between representation at both loci greater than 20-fold; and (iii) unusable, one or both loci with <0.1% representation as compared to genomic DNA. Samples falling into the highly usable category had a greater than 99.9% chance of being correctly assigned to a cluster in monoplex TaqMan genotyping assays, with a less than 1% chance of being uncallable (outside all genotyping cluster boundaries). Samples falling into the limited utility category had a greater than 99.0% chance of being correctly assigned to a cluster in monoplex TaqMan genotyping assays, with a less than 5% chance of being uncallable. Unusable samples fell below these standards. This algorithm was developed at Molecular Staging based on TaqMan-based genotyping analysis of thousands of amplified DNA samples from a variety of sources (data not shown). It should be noted that there may be amplified DNA sample sets or downstream genotyping assays for which this QA will not correctly predict the genotyping accuracy.
Sequence Analysis
All nonamplified genomic DNA and MDA-amplified DNA samples were diluted to 40 ng/µL for genotyping. For each sample, PCR amplification was carried out on 1 µL of diluted nonamplified genomic DNA and MDA amplified DNA in 25 µL reaction volumes. For some segments with high GC content, 5%–10% dimethylsulfoxide (DMSO) was added in the reaction. Before sequencing, PCR products were treated with exonuclease I and shrimp alkaline phosphatase (USB Corporation, Cleveland, OH, USA) for 30 min at 37°C and 15 min at 80°C to remove excess deoxynucleotide triphosphates and oligonucleotide primers. Sequencing reactions were performed with BigDye™ Terminator Cycle Sequencing Kit (Applied Biosystems). Samples were purified by extraction with Sephadex® G-50 (Amersham Biosciences, Piscataway, NJ, USA) and then run on ABI Prism 377 Automated Sequencer (Applied Biosystems). Sequence chromatograms were analyzed with Sequencher™ software (Gene Codes, Ann Arbor, MI, USA) for both genomic DNA and MDA-amplified DNA samples. Based on GC contents and availability of previously analyzed heterozygous polymorphisms, 15 common polymorphisms from 10 genes located on 8 chromosomes were examined using whole blood samples from 118 (115 with high-quality and 3 with lower quality MDA amplification) individuals (see Tables ).
Table 1. Overview of Analyses
Table 2. Summary of Genotyping for MDA-Amplified DNA
Table 3. Genotyping Common Polymorphisms for MDA-Amplified DNA Samples
Mutation Scanning
Mutation scanning was carried out for seven individuals from both genomic DNA and MDA-amplified DNA samples. Detection of virtually all mutations SSCP analysis (DOVAM-S), a multiplexed and highly redundant form of SSCP that detects virtually all mutations, was used for mutation scanning (Citation11). This variation of SSCP involves scanning of multiple exons in a single lane under five different nondenaturing electrophoresis conditions with different matrices, buffers, additives, and temperatures. For each patient, 22.8 kb of genomic DNA was scanned, including all exons and adjacent intronic regions for progesterone receptor (PR), estrogen receptor-β (ERβ), thyroid hormone receptor-β (TRβ), and vitamin D receptor (VDR) genes. Thirty-nine segments, ranging in size from 161 to 373 bp, were analyzed in three groups of 13 products spanning the size range. The segments were amplified robotically on the ABI Prism 877 Integrated Thermal Cycler (Applied Biosystems) and automatically pooled into a single tube. Following PCR amplification, the products were first electrophoresed on a denaturing acrylamide gel to detect PCR dropouts. PCR amplification was repeated for exons that did not amplify. PCR products were then electrophoresed on nondenaturing gels using five different electrophoresis conditions: (i) PagePlus with 30 mM Capso-ethanolamine, pH 9.6, at 4°C; (ii) PagePlus with 30 mM tricine-triethanolamine, pH 7.9, at 20°C; (iii) PagePlus with 50 mM Tris-borate, pH 8.3, 5% glycerol, at 20°C; (iv) HR1000 with 50 mM Tris-borate, pH 8.3, 2.5% glycerol, at 4°C; and (v) HR1000 with 30 mM tricine-triethanolamine, pH 7.9, at 4°C. Segments with mobility shifts were reamplified and sequenced on the 377 Automated Sequencer, and sequence chromatograms were analyzed with Sequencher software. Mutations were confirmed by reamplification from the original genomic DNA and sequencing in the opposite direction.
PCR of the Factor VIII Gene Inversion Segments
Subcycling PCR for the intron 22 inversion of the factor VIII gene was performed from MDA-amplified DNA and genomic DNA with four primers that differentiate the wild-type, inversion, and carrier (Citation12). Twelve- and ten-kilobase segments are produced in wild-type individuals. Males with hemophilia due to the inversion produce an 11-kb segment along with the 10-kb segment from the nonrecombined extragenic homolog. Female carriers have all the 10-, 11-, and 12-kb segments. In all cases, the 10-kb segment serves as an internal control. The conditions for subcycling PCR for the first 10 cycles were: 94°C for 12 s, followed by four subcycles of annealing/elongation that involve 60°C for 120 s and 65°C for 120 s for each subcycle (Citation12). The remaining 20 cycles were modified by the addition of an extra 3 s/cycle for each step of the annealing/elongation step. An additional 2 min of denaturation was utilized in the first cycle. Samples were electrophoresed on a 0.6% nondenaturing agarose gel and then stained with ethidium bromide for UV photography by a charge-coupled device (CCD) camera (Gel Doc™ 1000; Bio-Rad, Hercules, CA, USA).
Analysis of the Discrepant Samples
Sample 5071-2 was analyzed further. The absence of heterozygosity of the MDA-amplified DNA was confirmed by PCR reamplifying and resequencing. Four commonly used microsatellite markers, BAT-25, BAT-26, D2S123, and D17S250, were analyzed for amplified and nonamplified genomic DNA from this individual as a fingerprint of genetic identity (Citation13). The sample was resubmitted for amplification by MDA.
Chromatogram Analysis
The percentage of a sequence variant in the samples was assessed by the peak area ratio (PAR) computed by Phred software Version 0.020425.c (Citation14). Phred software calculates the area of each base in the sequencing chromatogram, and when examining heterozygote sequences, the peak area ratio of one allele to the other is fairly consistent within the sequence context of that allele. If one allele had not been amplified equally, this ratio would be different.
Results
A total of 252 samples analyzed with mutation scanning and direct sequencing for genomic variation associated with psychiatric disease were amplified by MDA. Many of the samples were extracted more than 10 years ago. It is not possible to obtain additional DNA from these patients. MDA-amplified DNA was analyzed by quantitative PCR TaqMan assays at two genomic loci by a quality control algorithm developed by Molecular Staging for predicting genotyping performance of the amplified DNA (see the Materials and Methods section). The MDA-amplified DNA was judged of poor quality (unusable) in two samples (0.79%) and of moderate quality (limited utility) in seven samples (2.8%). The remaining amplification products were of high quality (highly usable). One hundred fifteen samples with high-quality amplification were analyzed further. shows an overview of results. Samples were extracted with phenol/chloroform (Citation8), ATGC, or Puregene. DNA derived from three DNA extraction methods was amplified well by MDA.
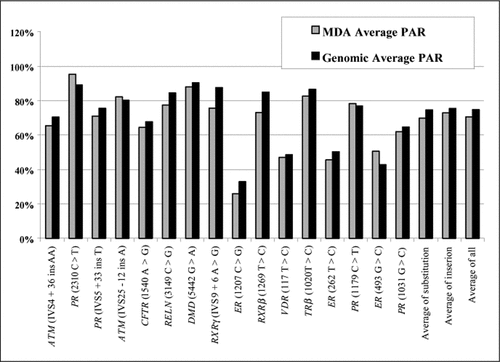
The input DNA for MDA ranged from 4 to 488 ng or about 1200-146,400 copies of the genome. Amplification products were quantified using the PicoGreen assay. DNA yields of postamplification products ranged from 481 to 821 µg with a mean concentration of 630 µg (average amplification equals 16,000-fold). To test whether MDA-amplified DNA is an accurate representation of the original genomic sequence, genotyping was performed by direct sequencing for 15 common polymorphisms located on 8 chromosomes. The relative peak heights from samples of genomic DNA and MDA-amplified DNA from known heterozygotes were compared. The GC content of these segments for genotyping ranges from 30% to 69% ( and 2). All 207 heterozygous loci were detected in high-quality MDA-amplified DNA samples. For the 15 polymorphisms, average PARs of the sequencing chromatograms of heterozygous samples for genomic DNA and MDA-amplified DNA were similar ().
Average peak area ratio (PAR) of the sequencing chromatograms for heterozygous samples of genomic DNA and multiple displacement amplification (MDA)-amplified DNA for the 15 polymorphisms were analyzed. PAR, area of the second peak/area of the first peak (as determined by Phred software). ATM, ataxia telangiectasia mutated; PR, progesterone receptor; CFTR, cystic fibrosis transmembrane conductance regulator; RELN, reelin; DMD, dystrophin (muscular dystrophy); RXRγ, retinoid X receptor-γ; ER, estrogen receptor; RXRβ, retinoid X receptor-β; VDR, vitamin D receptor; TRβ, thyroid hormone receptor-β.
PCR for Challenging Segments
To assess the utility of MDA for amplifying large exons for mutation analysis or for amplifying rather long regions for haplotype analysis, two particularly challenging segments of 10 and 12 kb, respectively, were chosen. These segments contain regions of varying GC content, from as low as 30% to a 3-kb region of more than 70%, including a 1-kb segment of about 80% GC. These large amplification products are required to detect the intron 22 inversion hotspot in the factor VIII gene, which is responsible for about 45% of severe hemophilia A. The inversion hotspot results from recombination between 9.4-kb virtually identical segments of the X chromosome that are separated by about 3 Mb.
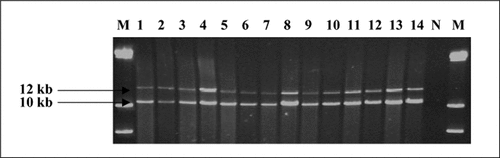
These segments are difficult to amplify, requiring special PCR conditions. The 10- and 12-kb segments of the human factor VIII gene region were successfully amplified in 20 of 20 MDA-amplified DNA samples. The 10-kb segment amplified with similar intensity while the 12-kb segment was amplified with lower intensity in the MDA samples relative to genomic DNA (). The underrepresentation of the 12-kb segment most likely reflects the efficiency with which this segment was amplified in MDA. This is in line with Dean et al. (Citation5) who estimated an average segment size of 10–12 kb.
PCR products were electrophoresed through a 0.6% agarose gel. The 12- and 10-kb PCR products from seven pairs of multiple displacement amplification (MDA)-amplified DNA and original genomic DNA are shown here. Lanes 1, 3, 5, 7, 9, 11, and 13 show PCR products of MDA-amplified genomic DNA. Lanes 2, 4, 6, 8, 10, 12, and 14 show PCR products of corresponding genomic DNA. N, no DNA control; M, λ DNA HindIII.
Candidate Gene Mutation Analysis
Mutation scanning by DOVAM-S, a highly redundant form of SSCP, was performed for seven patients for the PR, ERβ, TRβ, and VDR genes. The GC content of the amplified products ranged from 50% to 59%. For each patient, 22.8 kb of genomic DNA was scanned for mutation, including all coding regions and adjacent intronic regions for these genes. Thirty-nine segments were analyzed, ranging in size from 161 to 373 bp. A total of 159.4 kb of genomic DNA was scanned. Segments with mobility shifts seen on DOVAM-S gels were identical in the seven MDA-amplified DNA samples and their corresponding genomic DNA samples. Sequencing revealed the same variants in genomic DNA and MDA-amplified DNA samples ().
Table 4. Mutation Scanning in the Seven MDA-Amplified DNA Samples
Analysis of Lower Quality Samples
One sample with poor quality (unusable) and two samples with moderate quality (limited utility) were analyzed further by direct sequencing analysis. Four heterozygote and six homozygote loci were analyzed. Genotypes were identical for nonamplified genomic DNA and MDA-amplified DNA, except that one heterozygous allele was completely missed in one MDA sample (5071-2) of moderate quality. Genotype of nonamplified genomic DNA of this sample for the VDR gene was heterozygote (117 T/C, M1T), while after MDA, this sample was genotyped as homozygous C. Microsatellite analysis suggested that sample mix-up is unlikely. The sample was reamplified by MDA with a higher amount of input DNA (31 ng the first time versus 138 ng the second time), and both alleles were detected at the expected ratio. The quality of amplification was judged to be high by Molecular Staging.
Discussion
MDA-amplified DNA samples performed well in mutation sequencing, mutation scanning, and challenging PCR. Of particular interest for studies in molecular epidemiology is the accurate genotyping of heterozygotes. To our knowledge, this is the first analysis in which direct sequencing was used as a primary technique to examine heterozygosity in MDA-amplified DNA samples. As has been validated in this study, the MDA method is insensitive to local sequence (e.g., GC content and/or single nucleotide changes). While a very effective method, degraded DNA, poor quality extractions, or low amounts of template DNA may produce stochastic effects with respect to allele bias in the amplified DNA. When the input DNA amount was increased (from 31 to 138 ng) for the second MDA for the sample which had allele dropout, both alleles were amplified and detected in the expected ratio. While the first MDA-amplified DNA of this sample was scored as moderate quality, the reamplified DNA of this sample was scored with high quality. This highlights the importance of quality control. This was the only anomaly found in this extensive analysis.
The samples in the present study were amplified by an average of 16,000-fold. While the DNA amount varied substantially from sample to sample (more than 10-fold), the amount of DNA subsequent to amplification was very similar. Dean et al. amplified genomic DNA from 100- to 100,000-fold (Citation5). Other studies range from 2000- to 63,000-fold (Citation6,Citation7). One might expect a higher bias for larger amounts of amplification. More work is necessary to assess the performance of MDA as a function of the extent of WGA.
Templates with high GC content are particularly difficult to amplify, due to their high melting temperatures, and may require additional measures beyond optimizing reaction conditions. Incomplete separation of DNA strands can adversely affect amplification efficiency. In addition, template secondary structure or unmelted GC-rich regions can prevent primer binding and enzymatic elongation. To evaluate the effect of GC content on the fidelity of MDA, we genotyped 15 common polymorphisms from 10 genes located on 8 chromosomes with GC content varying from 30% to 69%. The results suggest that MDA-amplified DNA works efficiently in regions of high GC content.
The 10- and 12-kb segments of the factor VIII gene are difficult to amplify with PCR. Indeed, for many years after the descriptions of the common factor VIII inversion, molecular diagnosis was performed by Southern blot analysis, until special PCR conditions were developed (Citation12,Citation15). The ability to assay these segments in MDA-amplified DNA samples indicates that segments of widely varying GC content that are difficult to amplify by PCR can be amplified well by routine MDA conditions, supporting previous work by Detter et al. (Citation16) who cloned the MDA-amplified DNA and performed whole-genome shotgun sequencing.
For economic and technical reasons, it is often not possible to establish cell lines when patient samples are collected. Even if a few hundred micrograms of DNA are available, the resource is finite. Analysis of one candidate gene typically involves dozens and occasionally more than 100 PCR amplifications. MDA facilitates molecular analyses by generating an essentially infinite resource from moderate quantities of genomic DNA.
In conclusion, sequence analysis, amplification of rather long difficult DNA segments, and scanning-based candidate gene mutation analysis complement previous work and add to a growing body of evidence that WGA by MDA is a robust and accurate tool for genomic analysis. Quality control of the MDA-amplified DNA is very important. MDA has the potential to be utilized for noninvasive clinical testing (e.g., amplification of small amounts of DNA from buccal swabs). More work is necessary to determine if MDA meets the exact tolerances required for such clinical applications.
Acknowledgments
We thank Chunmei Yang and Jiesheng Chen for technical assistance, Dongqing Gu for chromatograms analysis, Christina Ticsay, Wenjia Song, and Ryan Quilatan for sequencing, Bita Estaki and Carolyn Buzin for the microsatellite analysis, and Xuemin Li and Hernanie Domingues for the assistance of FVIII intron 22 inversion PCR. This work was partially supported by U.S. Army BC990386.
References
- Cheung, V.G. and S.F.Nelson. 1996. Whole genome amplification using a degenerate oligonucleotide primer allows hundreds of genotypes to be performed on less than one nanogram of genomic DNA. Proc. Natl. Acad. Sci. USA93:14676–14679.
- Paunio, T., I.Reima, and A.C.Syvanen. 1996. Preimplantation diagnosis by whole-genome amplification, PCR amplification, and solid-phase minisequencing of blastomere DNA. Clin. Chem.42:1382–1390.
- Telenius, H., N.P.Carter, C.E.Bebb, M.Nordenskjold, B.A.Ponder, and A.Tunnacliffe. 1992. Degenerate oligonucleotide-primed PCR: general amplification of target DNA by a single degenerate primer. Genomics13:718–725.
- Zhang, L., X.Cui, K.Schmitt, R.Hubert, W.Navidi, and N.Arnheim. 1992. Whole genome amplification from a single cell: implications for genetic analysis. Proc. Natl. Acad. Sci. USA89:5847–5851.
- Dean, F.B., S.Hosono, L.Fang, X.Wu, A.F.Faruqi, P.Bray-Ward, Z.Sun, Q.Zong, et al.. 2002. Comprehensive human genome amplification using multiple displacement amplification. Proc. Natl. Acad. Sci. USA99:5261–5266.
- Hosono, S., A.F.Faruqi, F.B.Dean, Y.Du, Z.Sun, X.Wu, J.Du, S.F.Kingsmore, et al.. 2003. Unbiased whole-genome amplification directly from clinical samples. Genome Res.13:954–964.
- Tranah, G.J., P.J.Lescault, D.J.Hunter, and I.De Vivo. 2003. Multiple displacement amplification prior to single nucleotide polymorphism genotyping in epidemiologic studies. Biotechnol. Lett.25:1031–1036.
- Gustafson, S., J.A.Proper, E.J.Bowie, and S.S.Sommer. 1987. Parameters affecting the yield of DNA from human blood. Anal. Biochem.165:294–299.
- Ahn, S.J., J.Costa, and J.R.Emanuel. 1996. PicoGreen quantitation of DNA: effective evaluation of samples pre- or post-PCR. Nucleic Acids Res.24:2623–2625.
- Singer, V.L., L.J.Jones, S.T.Yue, and R.P.Haugland. 1997. Characterization of PicoGreen reagent and development of a fluorescence-based solution assay for double-stranded DNA quantitation. Anal. Biochem.249:228–238.
- Liu, Q., J.Feng, C.Buzin, C.Wen, G.Nozari, A.Mengos, V.Nguyen, J.Liu, et al.. 1999. Detection of virtually all mutations-SSCP (DOVAM-S): a rapid method for mutation scanning with virtually 100% sensitivity. BioTechniques26:932–942.
- Liu, Q. and S.S.Sommer. 1998. Subcycling-PCR for multiplex long-distance amplification of regions with high and low GC content: application to the inversion hotspot in the factor VIII gene. BioTechniques25:1022–1028.
- Boland, C.R., S.N.Thibodeau, S.R.Hamilton, D.Sidransky, J.R.Eshleman, R.W.Burt, S.J.Meltzer, M.A.Rodriguez-Bigas, et al.. 1998. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res.58:5248–5257.
- Ewing, B., L.Hillier, M.C.Wendl, and P.Green. 1998. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res.8:175–185.
- Liu, Q., G.Nozari, and S.S.Sommer. 1998. Single-tube polymerase chain reaction for rapid diagnosis of the inversion hotspot of mutation in hemophilia A. Blood92:1458–1459.
- Detter, J.C., J.M.Jett, S.M.Lucas, E.Dalin, A.R.Arellano, M.Wang, J.R.Nelson, J.Chapman, et al.. 2002. Isothermal strand-displacement amplification applications for high-throughput genomics. Genomics80:691–698.

