
Abstract
The persistence of donor cells in recipient circulation and peripheral tissues post-transplantation has been demonstrated in solid organ allotransplantation and xenotransplantation models. Although this state of microchimerism has been postulated as the basis for graft acceptance, chimerism has not been directly linked to the maintenance of peripheral tolerance or prevention of rejection. Studies have demonstrated that the qualitative presence or absence of donor microchimerism bears no association with graft acceptance. Our preliminary work suggests that there is a threshold chimerism necessary for the induction of donor-specific hyporesponsiveness. Because the kinetics of donor cell accumulation and distribution in allograft recipients are largely unknown, quantitative analyses are needed to evaluate chimerism's significance to donor-specific tolerance. We developed a quantitative, competitive PCR assay to precisely measure the amount of chimerism in male to female transplant pairs by targeting the sex-determining region of the Y chromosome (SRY gene). Traditionally, this technique requires that serial known amounts of an SRY-specific competitive template (CT) be coamplified with a constant amount of sample DNA to determine the equivalence point of the relative band intensities of the PCR products. However, running a panel of PCRs with CT amounts above and below the equivalence point to generate a standard curve for every sample is laborious. Here we describe the generation of a single standard curve that permits the rapid and reliable quantification of microchimerism after coamplification of sample DNA with a single amount of CT.
Introduction
Following the allotransplantation of solid organs, cells of donor origin persist in the peripheral circulation and tissues of the recipient. This phenomenon, termed donor microchimerism, is postulated to be paramount to the development of donor-specific hyporesponsiveness and graft acceptance (Citation1,Citation2). PCR amplification of the sex-determining region of the Y chromosome (SRY gene) is commonly used in male to female donor-recipient transplant pairs to detect persistent donor (male) cells in recipient (female) blood and tissues post-transplantation (Citation2–7). However, several studies have demonstrated that the qualitative presence or absence of donor microchimerism bears no association with clinical outcomes, including allograft acceptance (Citation8–12). Indeed, our preliminary results in a porcine model of intestinal transplantation indicate that a threshold level of microchimerism is necessary for the induction of donor-specific hyporesponsiveness (Citation13). Previous efforts to define the extent of microchimerism have typically employed variations of standard PCR methodology that permit, at best, the generation of semiquantitative data (Citation14). This inherent limitation of standard PCR methodology occurs because minute differences in any of the multiple variables that affect amplification efficiency can dramatically alter the product yield, thus precluding accurate quantification, and by extension, meaningful comparisons of levels of microchimerism between samples. Therefore, the development of a bona fide quantitative PCR assay, capable of determining the precise level of donor cells persisting within recipient tissues, is an essential technical prerequisite for the study of microchimerism. Fortunately, PCR can be adapted to provide truly quantitative information without sacrificing its high degree of sensitivity.
We developed a quantitative competitive PCR (QC-PCR) assay, based on our previously published methods (Citation15,Citation16), in which the unknown amount of native SRY DNA in the sample is coamplified with an SRY-specific competitor DNA template as an internal control. Because the same primers recognize both the target and the competitor template (CT), any variable influencing the amplification reaction similarly affects both the target and CT. Thus, the relative abundance of the coamplified products remains constant, even under different conditions of amplification efficiency. Although highly accurate, the necessity of running a panel of PCRs with CT amounts above and below the equivalence point to generate a graph (Citation15,Citation17,Citation18) for each individual sample is quite laborious and therefore impractical for the analysis of multiple samples. To address this problem, we have designed a more rapid, quantitative PCR technique to determine the degree of microchimerism in sex-mismatched transplant recipients. Our modified QC-PCR method is based on the generation of a standardized “master” curve.
In this report, we describe and validate a QC-PCR method whereby the generation of a single standard curve permits the degree of donor microchimerism to be precisely quantified in DNA samples extracted from a variety of recipient tissues.
Materials and methods
Primer and Competitor Design
Primers were designed to enable specific amplification of a 266-bp segment of the sex-determining region of the pig Y chromosome (SRY gene), 5′-AGTGGTAGAGAGAGTGGCCAGGATCGTG-3′ and 5′-TGTGCCCTCTCTCCCTTGCGACGAGGT-3′. The slightly longer CT was created by the insertion of a short DNA fragment (34 bp) into the native SRY gene sequence in a multistep, PCR-directed process using complementary internal oligoprimers (5′-GGCCCGCCCGGAACAAGAAGAACTGGGATGCAAGTGGAAAAT-3′ and 5′-CGGGCGGGCCATGATGTATAGGCCACTTGCTGATCTCTGAGT-3′). The 300-bp CT was then ligated into the TOPO TA Cloning® Vector (Invitrogen, Carlsbad, CA, USA), a plasmid clone containing the CT insert was extracted from an overnight bacterial culture with a Wizard® Plus Miniprep DNA Purification System (Promega, Madison, WI, USA), and the DNA yield was measured by a spectrophotometer. A stock solution of 1 ng/µL of CT was prepared, from which the desired dilutions were obtained.
DNA Amplification
DNA was extracted from 0.5–1.0 g of tissue or 15 cm3 of peripheral blood, according to the methods of Maniatis et al. (Citation19), using phenol-chloroform and ethanol precipitations. Chimeric DNA samples (1000 ng) were prepared by mixing male genomic DNA with female genomic DNA to achieve final male DNA concentrations ranging from 10% to 0.001%. Serial dilutions of CT were prepared from the stock solution (amounts ranging from 1 to 0.0000001 pg). PCR was carried out in 50-µL reaction mixtures, including 1.2 µM (80 ng) each of sense and antisense primers, 200 µM dNTP, 2 mM MgCl, 2.5 U of Taq DNA polymerase, and 5 µL of 10× reaction buffer (750 mM Tris-HCL, pH 8.8, 200 mM (NH4)2SO4, 0.1% Tween® 20). Amplification was performed in a Perkin Elmer Gene-Amp® 2400 Thermal Cycler (Applied Biosystems, Foster City, CA, USA), with a 7-min denaturation at 94°C followed by 35 cycles of 15 s at 94°C, 15 s at 65°C, 30 s at 72°C, and a final 10-min extension at 72°C. PCR products (10 µL) were then resolved by electrophoresis on a 2.5% agarose gel (Roche Applied Science, Montreal, QC, Canada) stained with ethidium bromide.
Generation of Standard Curve
The SRY primers were used to coamplify male DNA and CT. The amount of male genomic DNA used was held constant at 0.1 ng (equivalent to 15 diploid cells or copies of the SRY gene) admixed with female DNA for a total of 1000 ng chimeric DNA (0.01% male DNA) per reaction tube, while the CT varied over a range from 0.033 to 0.00001 pg in 11 separate reactions. The PCR products were resolved on a 2.5% agarose gel and scanned under ultraviolet (UV) light using the Alpha Imager 2000 Digital Imaging System (Alpha Innotech, San Leandro, CA, USA), with careful attention paid to the avoidance of band saturation.
Transplantation Model
Orthotopic small bowel transplantations using male donor and female recipient outbred pigs were performed as previously described (Citation13). Blood samples were collected at interval days post-transplantation, and tissue samples were procured at autopsy.
Results and discussion
Our QC-PCR method relies on the coamplification of the native SRY gene from pig genomic DNA and an artificial variant containing a 34-nucleotide insertion (the CT). The absolute minimum limits of detection for both the SRY DNA and the CT were first individually determined following the optimization of PCR conditions. The limits of detection were found to be a concentration of 0.0003% (male to female ratio of 1:3 × 105) for the SRY gene and a dilution of 0.000001 pg for the CT, respectively. We then performed a titration with a constant amount of male genomic DNA (0.1 ng in a 10,000-fold excess of female DNA) and known amounts of the CT. When the amplifications were analyzed by agarose gel electrophoresis, as expected, both the 266-bp band representing the SRY DNA and the 300-bp band corresponding to the amplified CT product were visualized (). A standard curve was generated by plotting the log of the CT/SRY band intensity ratio on the y-axis against the log of CT dilutions in picograms on the x-axis (). The observed linearity (r2 = 0.992) indicates a constant relationship between the SRY and CT amplification efficiencies (Citation20). The regression equation of the line is y = 0.6605x + 2.9805. Because the amplification kinetics of both CT and SRY DNA are highly similar, it is valid to conclude that equal PCR product band intensities can occur only when the starting amounts of CT and SRY substrate are identical. Therefore, when CT/SRY band intensities are equal (y = 0), the antilog of x gives the starting amount of CT prior to amplification, which is also the starting amount of the SRY DNA. Applying the equation in this manner, the calculated amount of SRY DNA in 100 pg of male genomic DNA interpolated from this plot is 0.0000307 pg (0.0000307%), with a coefficient of variation (cv) of 7.31% on three replicate experiments.
(A) Agarose gel electrophoresis of the coamplification of 0.1 ng of male genomic DNA (male DNA mixed with female DNA at a ratio of 1:104) with serial competitive template (CT) amounts ranging from 0.033 to 0.00001 pg. The gel shows two PCR products per lane, representing the CT (larger fragment, 300 bp) and SRY gene (smaller product, 266 bp). (B) Standard curve generated from the gel shown in panel A. The band intensity is assayed by scanning densitometry, and the graph is generated by plotting the log CT/SRY band intensity against the log CT amount. (C) Standard curves generated from amplifications containing three initial concentrations of male DNA substrate. Standard curve (i) represents the data from panel B (0.1 ng male DNA); (ii) and (iii) correspond to 1 and 10 ng male DNA, respectively.
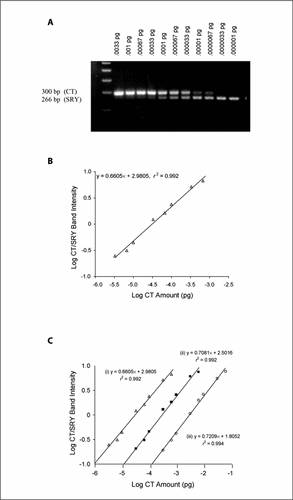
To assess the internal consistency of QC-PCR kinetics over a wide range of starting amounts, two additional experiments were performed using 1 and 10 ng of male genomic DNA admixed with 1000 ng of female DNA as substrate. These mixtures, representing 0.1% and 1% male DNA content, were coamplified with CT amounts ranging from 0.01 to 0.00001 pg and 0.1 to 0.0001 pg, respectively. The linearity of coamplification was confirmed, as indicated by the respective r2 values of 0.992 and 0.994. The SRY amounts in picograms were calculated using the respective linear equations generated by each graph (when y = 0, antilog of x = CT amount prior to amplification = SRY amount in picograms; ). SRY amounts of 0.000293 and 0.00313 pg were detected using 1 and 10 ng of male DNA, respectively. Expressing these values as the percent of male genomic DNA occupied by the SRY gene yields values of 0.0000293% and 0.0000313%, results that are highly consistent with the initial determination of 0.0000307%.
Thus, the kinetics of amplification between the CT and SRY substrates remained constant over a 2 log-fold range of starting amounts. This vital property of QC-PCR permits the use of a single master standard curve to quantify the male DNA concentration in multiple blood and tissue samples obtained from female chimeras post-transplantation. Moreover, once this master curve has been generated, only a single QC-PCR, yielding both a CT and SRY product, needs be performed.
To test the practical application of our master standard curve methodology, we analyzed liver tissue harvested from a female recipient of a male intestinal graft for the degree of donor microchimerism. As a basis of comparison, we first determined the amount of SRY DNA in the sample using the conventional approach of coamplification of SRY DNA in the sample with various dilutions of CT (0.0001, 0.00001, and 0.000001 pg), followed by the generation of a standard curve (, A and B). The resultant linear regression equation (y = 0.5871x + 2.8543) was then used to calculate the amount of SRY in the usual fashion (when y = 0, antilog of x = SRY amount in picograms). The amount of SRY DNA present in the tissue sample by this method was determined to be 0.0000137 pg. Because we have previously demonstrated that the SRY gene occupies 0.0000307% of male genomic DNA, we can calculate a total of 44.625 pg of male genomic DNA in the sample. Furthermore, because 1000 ng of DNA were harvested from the liver of the recipient female in this experiment, the degree of donor microchimerism can be expressed as 0.0045%.
(A) Electrophoresis of 1000 ng of DNA extracted from a female liver post-transplantation submitted to standard PCR with SRY-specific primers and coamplified with competitive template (CT) of (Citation1) 0.0001, (Citation2) 0.00001, and (Citation3) 0.000001 pg, respectively. Scanning densitometry of the product bands generated ratios of 3.20, 0.83, and 0.21, respectively. (B) A conventional standard curve of the data shown in panel A, in which data are plotted as the log CT/SRY band intensity ratio versus the amount (amt) of CT used. (C) The master standard curve, generated from the conventional standard curve in panel B by the inclusion of male DNA amount added to the denominator on the x-axis.
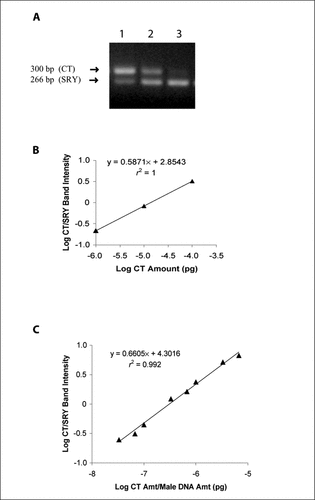
We then applied our master standard curve methodology to calculate the percent of microchimerism in the same liver tissue sample. Because we have shown that the kinetics of amplification for CT and SRY DNA remains constant over a wide range of starting substrates, any of the conventional standard curves depicted in can be transformed into the master standard curve. The selected standard curve is transformed by replotting the data points obtained from the competitive PCR using a modified x-axis that reflects the amount of male DNA used as substrate in the original serial coamplifications. For the standard curve depicted in , this modification produces the master standard curve that is defined by the new regression equation y = 0.6605x + 4.3016, where y = log (CT/SRY) band intensities and x = log [CT amount (pg)/male DNA amount (pg)] (). This regression equation can subsequently be used to calculate the amount of male DNA in any tissue sample procured from a female chimera because the only unknown in this equation is the male DNA amount (pg), and the only requirement is a single coamplification of the sample with a CT amount that yields both SRY and CT PCR products. Scanning the middle lane of the gel in , which resulted from using 0.00001 pg of CT substrate in the PCR, yields a CT/SRY band intensity ratio of 0.83. Substituting the appropriate values into the regression equation gives log 0.83 = 0.6605 log [0.00001 pg/male DNA amount (pg)] + 4.3016. Solving for the male DNA amount gives a result of 43.16 pg, equivalent to 0.0043% male DNA in the 1000 ng of liver tissue. Thus, the master standard curve methodology yields a virtually identical degree of microchimerism to that obtained by the conventional method, without the need for multiple coamplifications for each sample analyzed.
In total, we performed 26 small bowel transplants, and DNA was extracted not only from liver but also from spleen, thymus, and skin procured at autopsy, in addition to blood samples collected at select intervals postoperatively. The SRY amount was calculated for each sample using both the conventional standard curve technique and the new master curve strategy. Altogether, in excess of 100 direct comparisons were made, yielding virtually identical results each time (cv < 8). In , representative data from two animals are presented, showing that the master standard curve method reliably quantifies the male donor microchimerism in native recipient female tissues, independent of the source of DNA (tissue type) and experimental condition.
Table 1. Comparison of Conventional Standard Curve and Master Standard Curve Applications for Quantitative Analysis of Male DNA in Various Tissues
One potential critique of our methodology is that since the slope of the standard curve reflects the relative efficiencies of amplification of the SRY and CT templates, ideally, a standard curve with a slope of 1 would be generated (Citation20). However, this painstaking exercise would require the testing of many different CT to find one with identical amplification efficiency to that of the target DNA substrate and is clearly unnecessary for most practical applications. Our standard curve resulted in a slope less than 1, indicating a higher amplification efficiency of the SRY substrate compared to the CT. Therefore, although the amount of starting SRY substrate interpolated from the curve is precisely quantified, it has not been corrected for differences in amplification efficiency between the CT and SRY substrates. Nonetheless, the constant amplification kinetics between the SRY gene and CT permits reliable, quantitative comparisons of the levels of donor microchimerism between samples, both between recipients and from an individual recipient over time. Thus, this technique fulfills the necessary methodological prerequisites for the analysis microchimerism and thereby allows a meaningful assessment of the kinetics, extent, and impact of microchimerism on both recipient immunological function and on the experimental means used to achieve it.
Chimerism is a very dynamic process, with donor DNA concentrations in native recipients that can vary greatly both between tissues and over time. It is therefore highly desirable that a quantitative assay using a single standard curve be able to accurately determine levels of microchimerism over this entire range. By virtue of the constant amplification kinetics between the SRY and the CT demonstrated in this paper over a wide range of male DNA starting concentrations, the standard curve methodology that we describe here permits rapid, sensitive, and reliable quantification over the whole range of microchimerism using a single PCR. This methodology facilitates the analysis of kinetics of microchimerism post-transplantation with respect to outcomes and regimens implemented in any experimental setting where male-to-female transplant pairs are used.
Real-time PCR has emerged as a precise and reproducible technique for the quantification of gene expression that may be more rapid and efficient than the application we propose. However, real-time PCR is neither affordable nor accessible to most individual investigators. Furthermore, although the design of the master standard curve may appear time-consuming and cumbersome at the outset, with real-time PCR, the design of primers and probes and the optimization conditions also require a large investment of time and a lot more money. As such, the gel-based QC-PCR technique, using the master curve strategy described in this paper, is a reliable, sensitive, and cost-effective approach for the individual investigator needing to quantify gene expression rapidly, while avoiding the consumption of scarce amounts of DNA.
Acknowledgments
Nancy Morin is supported by a project grant from the American Society of Colon and Rectal Surgeons. Mark Lipman is a recipient of a senior clinician-scientist award from the Fonds de la Recherche en Sante du Quebec. This work was supported by the Kidney Foundation of Canada.
References
- Starzl, T.E., A.J.Demetris, N.Murase, S.Ildstad, C.Ricordi, and M.Trucco. 1992. Cell migration, chimerism, and graft acceptance. Lancet339:1579–1582.
- Starzl, T.E., A.J.Demetris, M.Trucco, N.Murase, C.Ricordi, S.Ildstad, H.Ramos, S.Todo, et al.. 1993. Cell migration and chimerism after whole-organ transplantation: the basis of graft acceptance. Hepatology17:1127–1152.
- Tashiro, H., Y.Fukuda, A.Kimura, S.Hoshino, H.Ito, and K.Dohi. 1996. Assessment of microchimerism in rat liver transplantation by polymerase chain reaction. Hepatology23:828–834.
- Inman, B., B.Halloran, A.Melk, V.Ramassar, and P.F.Halloran. 1999. Microchimerism in sensitized renal patients. Transplantation67:1381–1383.
- Zhang, J., K.L.Tong, P.K.Li, A.Y.Chan, C.K.Yeung, C.C.Pang, T.Y.Wong, K.C.Lee, et al.. 1999. Presence of donor- and recipient-derived DNA in cell-free urine samples of renal transplantation recipients: urinary DNA chimerism. Clin. Chem.45:1741–1746.
- Olszewski, W.L., M.Durlik, B.Lukomska, P.Religa, H.Ziolkowska, S.Janczewska, E.Cybulska, J.Soin, et al.. 2000. Donor DNA can be detected in recipient tissues during rejection of allograft. Transpl. Int.13:S461–S464.
- Tajik, N., D.Singal, G.Pourmand, M.Ebrahimi-Rad, M.Radjabzadeh, P.Tavasoli, F.Khosravi, and B.Nikbin. 2001. Prospective study of microchimerism in renal allograft recipients: association between HLA-DR matching, microchimerism and acute rejection. Clin. Transplant.15:192–198.
- Elwood, E.T., C.P.Larsen, D.H.Maurer, K.L.Routenberg, J.F.Neylan, J.D.Whelchel, D.P.O'Brien, and T.C.Pearson. 1997. Microchimerism and rejection in clinical transplantation. Lancet349:1358–1360.
- Ishida, H., T.Kawai, K.Tanabe, Y.Hayasaka, M.Yasuo, H.Toma, and K.Ota. 1996. Status of microchimerism in recipients 15 years after living related kidney transplantation. Transplantation62:126–128.
- Schlitt, H.J., J.Hundrieser, M.Hisanaga, K.Uthoff, M.Karck, M.Achtelik, T.Wahlers, K.Wonigeit, et al.. 1995. Donor-type microchimerism after heart transplantation—a dynamic process. Transplant. Proc.27:155–157.
- Suberbielle, C., S.Caillat-Zucman, C.Legendre, C.Bodemer, L.H.Noel, H.Kreis, and J.F.Bach. 1994. Peripheral microchimerism in long-term cadaveric-kidney allograft recipients. Lancet343:1468–1469.
- Ueda, M., J.Hundrieser, M.Hisanaga, K.Tanaka, K.Wonigeit, R.Pichlmayr, H.J.Schlitt, et al.. 1997. Development of microchimerism in pediatric patients after living-related liver transplantation. Clin. Transplant.11:193–200.
- Morin, N., E.Lamoureux, M.L.Lipman, J.Tector, J.I.Tchervenkov, and P.Metrakos. 2000. Donor-specific portal transfusions augments microchimerism in a porcine model of small bowel transplantation. Transplant. Proc.30:1290–1291.
- Terakura, M., N.Murase, A.J.Demetris, Q.Ye, A.W.Thomson, and T.E.Starzl. 1998. Lymphoid/nonlymphoid compartmentalization of donor leukocyte chimerism in rat recipients of heart allografts, with or without adjunct bone marrow. Transplantation66:350–357.
- Lipman, M.L., A.C.Stevens, and T.B.Strom. 1994. Heightened intragraft CTL gene expression in acutely rejecting renal allografts. J. Immunol.152:5120–5127.
- Strehlau, J., M.Pavlakis, M.Lipman, M.Shapiro, L.Vasconcellos, W.Harmon, and T.B.Strom. 1997. Quantitative detection of immune activation transcripts as a diagnostic tool in kidney transplantation. Proc. Natl. Acad. Sci. USA94:695–700.
- Becker-Andre, M. and K.Hahlbrock. 1989. Absolute mRNA quantification using the polymerase chain reaction (PCR). A novel approach by a PCR aided transcript titration assay (PATTY). Nucleic Acids Res.17:9437–9446.
- Gilliland, G., S.Perrin, K.Blanchard, and H.F.Bunn. 1990. Analysis of cytokine mRNA and DNA: detection and quantitation by competitive polymerase chain reaction. Proc. Natl. Acad. Sci. USA87:2725–2729.
- Maniatis, T. 1982. Molecular cloning. CSH Laboratory Press, Cold Spring Harbor, NY.
- Freeman, W.M., S.J.Walker, and K.E.Vrana. 1999. Quantitative RT-PCR: pitfalls and potential. BioTechniques26:112–125.

