
Free Expression
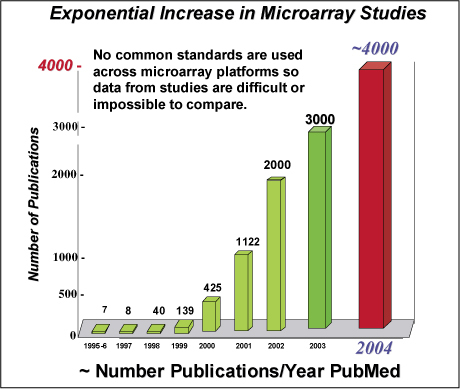
Authors of papers describing new gene or genome sequences have been expected for some years now to deposit their primary data in permanent, public data repositories, which include databases maintained by DNA Data Bank of Japan (DDBJ), European Bioinformatics Institute (EBI), and the National (U.S.) Center for Biotechnology Information (NCBI). Since its establishment in 1999, the Microarray Gene Expression Data Society (MGED) has suggested that all scientific journals should, as a condition of publication, require authors to submit their microarray data to public repositories in a similar fashion. Currently, there are three independent repositories: Gene Expression Omnibus (GEO) at NCBI, ArrayExpress at EBI, and Center for Information Biology Gene Expression Database (CIBEX) in Japan. Ron Edgar of NCBI believes researchers may find GEO particularly useful, as it makes their data visible by providing links to their publications and their sequence and/or genomic information. MGED further proposes that the primary responsibility for submission of data lies with the authors and that the databases work with both authors and journals to establish data submission and release protocols to assure compliance with their guidelines. BioTechniques expects to be fully compliant with MGED proposals by the beginning of 2005. Given the ever increasing number of studies using microarrays to analyze gene expression, standards for controls as well as data are much needed.
Data Standards
MGED has established MIAME (Minimum Information about a Microarray Experiment) guidelines for defining the content and structure of data from microarray-based studies. The MIAME checklist provides the means for authors to make certain they are providing sufficient information to allow others to analyze their data. During the review process of submitted papers, journals will need not only to enlist reviewers who are qualified to assess the data and determine if these guidelines have been met, but also provide secure, confidential prepublication access to those data.
MGED believes that the increasing availability of software to facilitate submission of microarray data directly to databases will relieve the considerable burden of this activity. Part of these standards include how the raw data are analyzed and transformed, as these activities significantly affect the results of and conclusions drawn from the experiments. As John Quackenbush, The Institute for Genomic Research, notes, “for DNA sequence data, the tissue source, age of the individual, treatment with drugs, presence of stress, all don’t matter. DNA is DNA. But all of those conditions and more affect RNA and its level of expression, so how to collect and capture data is challenging. Now that the genome project is well underway, the next big data set will consist of expression profiles and databases which will be very different from the linear,fixed genome project data.”
Neil Winegarden, University Health Network, Toronto, and local organizer of the 7th International MGED Meeting observes, “most people want to be compliant with the standards, but need information on how to do it and what resources are available.” The meeting provided tutorials for MGED, with information about each data repository including how to submit data. He hasn’t encountered anyone, particularly among the meeting population of academics, who are resistant to the overall goals of MGED. For those in industry and drug discovery, the limitations on publishing do not seem to affect submission of microarray data, since once they have permission to publish results, they usually have permission to make the array data available as well. His one area of concern is that there may be a delay in making data available on the part of individuals who feel they are not through mining their data, but he does not see this as a prevalent problem. In theory, one might choose to publish a microarray data-based article in a noncompliant journal to avoid disclosing the data, but since most journals, including all the prominent ones, require compliance, this gambit, if even possible, may be self-defeating.
Experimental Standards
The problems of comparing microarray data go beyond the creation and archiving of databases. What those data represent can be an issue, as microarrays do not measure gene expression in any quantitative units. The recommendation to develop microarray controls came out of a joint National Institute of Standards and Technology (NIST)—Stanford University meeting, Metrology and Standards, Needs for Gene Expression Technologies: Universal RNA Standards, in March 2003. Marc L. Salit, Research Chemist, Chemical Science and Technology Laboratory at NIST, is working with Janet A. Warrington, Vice President, Emerging Markets and Molecular Diagnostics Research & Development at Affymetrix to coordinate an industry-led effort to develop RNA standards for gene expression platforms, the External RNA Control Consortium (ERCC). Participants in ERCC include numerous private sector, public sector, and academic organizations, including the American Type Culture Collection, the Food and Drug Administration (FDA), National Cancer Institute (NCI), National Institute of Allergy and Infectious Disease, and NIST. The proposed external RNA controls are intended to be spiked into any gene expression platform (microarray or quantitative RT-PCR) to evaluate the technical performance of an experiment. These platform-independent controls should allow validation of the robustness of a gene expression experiment by comparing the observed to expected response to the spike-in dose. In addition to producing the controls, ERCC will also be developing a base set of informatics tools that permit performance evaluation and will generate protocols for their use. These materials and tools will be readily accessible, with all information and validation data published in the open literature. The informatics tools will be developed and made available in an open source format. These controls are being designed for use in both research and clinical applications. The controls will consist of about 100 well-characterized RNA transcripts derived from random unique sequences (as determined by comparison with mouse, rat, human, Drosophila, Escherichia coli, and mosquito sequence databases), and with well-characterized clones of Bacillus subtilis and Arabidopsis thaliana. A set of well-characterized pools with the same transcripts in different concentrations across pools will be developed for spiking into total RNA.
Ernest S. Kawasaki, Head of the Advanced Technology Center Microarray Facility at NCI, whose group is part of ERCC, says the easy part is the technical part and that it will be harder to get all the companies together to sell the controls in a noncompetitive fashion. A lot of the controls have been assembled, with testing to begin in early 2005. Later in 2005, a meeting is planned where the decision will be made on a set of controls good enough for public use. He expects a 3–5 year time frame tofinish the project. This should help make raw data available for cross-laboratory analysis. “We see good correlation across platforms, but you can’t overanalyze the data, it’s qualitative,” he notes. “You need quantitative PCR to get an absolute amount of expression.” As far as differences in what is on one chip in one lab versus another, there is no way to get same results, but one should get same type of answer, up-regulation or down-regulation. “Common standards,” he concludes, “will make it easier to compare.”
The FDA’s interest in ERCC results from the increasing use of microarray data in the development of diagnostics, in pharmacogenetic analyses, in preclinical and population clinical studies, and as support for claims of safety or efficacy. From their perspective, it’s dif?cult if not impossible to compare microarray data if all experimenters use their own sets of controls. Variability can arise from the quality of starting material, how it’s labeled or amplified, the stringency of hybridization, type of probes (e.g., oligonucleotide versus cDNA), the types of primers [e.g., oligo(d)T versus random hexamers], and how the image that is obtained is scanned, as well as variations in instrument settings among users of the same technology.
Courtesy of Ernest S. Kawasaki, Advanced Technology Center, NCI
Future Directions
Winegarden notes that with three independent microarray data repositories, some mechanism for cross-talk between them will need to be implemented in the near future. Theoretically, one could download data into one’s own system to analyze, but there is no easy read-through from one database to the others. Quackenbush predicts the future will bring a requirement for standards similar to MIAME for proteomics, “metabolomics,” in situ data, and toxicology data among others.
Courtesy of Ron Edgar, NCBI
Kawasaki predicts microarray analysis will allow for tailored treatment for all cancers. For many cancers, the subtype of disease cannot be distinguished by looking at cells, but requires some sort of molecular markers, be it gene expression patterns or antibody reactivity. He estimates that there will probably be fewer than 100 genes that will be important, and within perhaps 10 years, diagnosis by checking for specific markers will be in widespread use. Now that the “final” number of genes from human genome project is 20,000 to 25,000 genes, it is fairly easy to analyze on one chip. Laser capture to isolate highly purified tumor cells from a tissue sample allows one to determine which genes are expressed. He expects to see a limited number of common tumor-specific genes, so tumor diagnostic chips may need only a small number of genes even for disease-specific diagnosis as well as for determining appropriate therapy. Tumor characterization is possible now, but it’s still expensive, although Kawasaki expects within 5 years the cost will be much more reasonable. As an example, he observed that the cost now to print 30,000 genes on an array is $50, whereas several years ago the cost was in the many thousands of dollars.
writer in Mamaroneck, NY.

