

Abstract
Mass spectrometry has been used for decades and continues to be an integral part of analytical research. This feature explores its latest applications.
Just over 100 years since the development of the first mass spectrometer by Francis Aston (University of Cambridge, UK), the technique of mass spectrometry (MS) remains, to this day, a vital tool in the arsenal of researchers throughout the multifaceted field of life science [Citation1]. The modern-day spectrometer exists in a form that Aston would unlikely recognize and can now perform tasks that must surpass the wildest aspirations of the technology's pioneer.
Developments to the ionization, flight path and analysis protocols in MS have helped keep the spectrometer a relevant and valuable piece of equipment. Key examples of these developments include electrospray ionization, which has enabled the conversion of analytes from solution into the gas phase without too much disruption to the natural conformation of the molecule. This has led to an increase in the importance of the mass spectrometer in the field of structural biology, where it can now provide valuable information on the structure of proteins and other biological molecules in their native state – a technique known as native MS (nMS).
Technological developments in nMS for structural biology
One such recent development in the process of nMS is the combination of the technique with surface-induced dissociation (SID) – a high-energy method of fracturing native molecules in the gas phase [Citation2].
A team of researchers from Ohio State University (OH, USA), led by Vicki Wysocki and Steffen Lindert, identified that SID could be used to study the complex structure and binding behaviors of protein complexes in a mass spectrometer [Citation3].
Typically, structural biologists aim to glean structural and binding information on protein complexes while they are intact. Wysocki and Lindert, however, identified that by using SID they could flip this entrenched system of thought on its head, instead observing the complexes as they are split apart and examining how they divide to establish information regarding their structure, binding and subunit interaction.
When ionizing samples to the gas phase, the team subjected the native structures of the molecules to SID, which involves colliding the ionized molecules with a surface, as opposed to using traditional collisional activation techniques that rely on the ions colliding with buffer gas molecules. SID provides more rapid, higher energy impacts that lead to complex shattering into intact subunits, as opposed to slowly unfolding subunits into their secondary structures until they are released [Citation2].
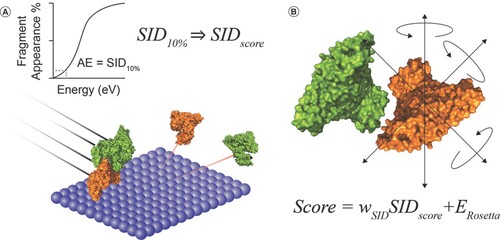
Using this method to separate the complexes, the team was able to generate mass spectra data from the fully folded subunits, which they used to identify a quality that they termed SID appearance energy (AE). AE is defined as 10% fragmentation of the complex (). Combining this experimental approach with the Rosetta modelling environment to create a ‘SID Score’ enabled the researchers to infer key pieces of information regarding the nature of the protein–protein interactions and poses at which the fragmentation takes place; for instance, subunit rigidity and the number of unsatisfied H-bonds () [Citation3].
(A) A depiction of surface-induced dissociation (SID) and a graphical representation of the generation of SID appearance energy. (B) A molecule model generated in Rosetta and the Scoring function equation.
This is but one key example of the ways nMS is developing in order to uncover new and vital information within structural biology. While technological developments continue to improve the mass spectrometer, the applications of the technique widen.
Using nMS to explore glycosylation
Post-translational modification (PTM) of proteins is a well-established and staggeringly abundant phenomenon. Of all the PTMs to occur in the domain of Eukaryota, glycosylation is the most prevalent, modifying almost half of the human proteome. Due to this abundance, the impact of its dysfunction is catastrophic, while the need to clearly understand this process is clear [Citation4].
As a result of the flexibility and heterogeneity of oligosaccharides, more established methods of structural examination, such as cryo-electron microscopy and x-ray crystallography, are ineffective. nMS, meanwhile, is fully capable of examining the extent and type of glycosylation present in such molecules as IgG, as mass shifts can be detected with a high enough resolution to correlate them to individual hexose rings [Citation5].
This ability recently enabled researchers from the University of Oxford (UK), led by the world-renowned practitioner of nMS, Carol Robinson, to establish the effects of glycosylation on the drug–protein interactions between warfarin and the serum protein alpha-1-acid glycoprotein [Citation6].
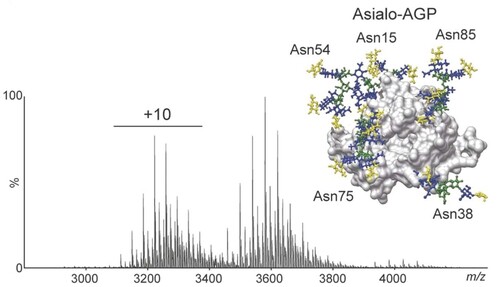
By conducting analyses of native mass spectra of the alpha-1-acid glycoprotein (), Robinson and her team found that elevated N-glycan branching of the glycoprotein, alongside terminal fucosylation, led to a decreased binding affinity of the glycoprotein to warfarin. This information provides an indication of how glycosylation can affect drug–protein interactions and, in turn, reveals useful insights about drug transport in the blood [Citation6].
Blue: GlcNAc; Green: Man; Yellow: Gal.
This example of nMS's high resolving power not only demonstrates the versatility of the technique, but also highlights its potential application in the study of disease and drug discovery.
nMS in microbiology
The refinement of MS has not only allowed for the analysis of viral and bacterial proteins in the native state, but advances that have reduced the limitations imposed on the technique by the size of the analyte have now enabled the ionization of full-size virions and capsids into the spectrometer for analysis [Citation7].
This advancement in technology has provided microbiologists with a whole new tool to analyze and explore aspects of virology. One such example of this is the recent study by Nyiri et al., which explored the UTPase family of proteins encoded by the genome of phage () that target Staphylococcus aureus. These UTPases can be dimeric or trimeric, and have previously been shown to engage with the S. aureus repressor protein StI, impacting the bacterium's pathogenicity island SaPIbov1 in the S. aureus genome [Citation8].
The team decided to explore the interactions between the UTPases, identifying their properties while also documenting the nature of the binding mechanism between the StI protein and the dimeric φNM1 phage dUTPase. To accomplish this, a combination of nMS, cross-linking and hydrogen deuterium exchange-MS experiments were employed [Citation8].
The team were able to identify that the StI protein is capable of displaying disparate stoichiometry and regions of peptide sequences depending on which phage UTPase it was interacting with, indicating the functional plasticity of StI. The nMS data were particularly vital in demonstrating that the mechanism by which StI interacted with and inhibited the dimeric UTPases was due to the architecture of the active site of the protein, which resides at the dimer binding site of the protein [Citation8].
Just as nMS has evolved to accommodate the analysis of larger molecules, as exemplified by its increasing use in virology, the increased acuity and resolution of the technique allow for the study of more delicate molecules than proteins, such as nucleic acids.
nMS in the study of nucleic acids
The application of MS in the field of genomics has previously included the detection of single-nucleotide polymorphisms and short tandem repeats; an application enabled by the development of matrix-assisted laser desorption/ionisation and electrospray ionization techniques [Citation9].
However, a recent paper published in Analyst describes the application of nMS to study the structure of telomeric G-quadruplexes [Citation10].
G-quadruplexes are structures formed at the end of strands of DNA and RNA in the telomeres that help protect telomeres from degradation, a process vital to the safeguarding of chromosome integrity and, consequently, cell proliferation. The polymorphic nature of these structures leads to difficulty studying their conformation using typical structural biology techniques [Citation10].
Speaking to study co-author Valérie Gabelica at the inaugural Celebration of Native Mass Spectrometry (Oxford, UK, 24–26 March 2019) while this research was being conducted, Gabelica noted that, “There are some aspects of the folding that we can reveal quite readily with MS, and which are difficult to get a hint of with traditional biophysical techniques” [Citation11].
With this in mind, Gabelica and co-author Valentina D'Atri set out to employ native ion mobility MS (IM-MS) to characterize the structure of different G-quadruplex topologies and to reveal aspects of their interactions such as cation binding and multimer formation in both four and eight repeat sequences of DNA and RNA.
The researchers found that in 8-repeat sequences of DNA, subunit folding of different topologies are linked – the formation of one subunit inhibits the folding of a second – while in RNA the subunits fold collaboratively via cation mediated stacking [Citation10].
This structural knowledge could prove valuable when trying to understand conditions of aging or chromosomal damage and clearly extolls the power of nMS in structural biology.
Exploring evolution
The direct application of nMS to pathogenic and human molecules and complexes in order to discover new avenues for research into our health and to identify targets for drug discovery could result in benefits for our future. Looking deep into the evolution of specific complexes, however, could also prove of significant value.
With this in mind, a recent study led by Michal Sharon of the Weizmann Institute of Science (Rehovot, Israel) used nMS techniques to study the evolutionary history of the 20S proteasome. This proteolytic complex is ubiquitous throughout all three kingdoms of life, making it an ideal candidate to study throughout evolution.
20S proteasomes were isolated from yeast, rat liver and human cells, all of which were reported in a paper published in Biomolecular Engineering, although in a presentation at the Celebration of Native Mass Spectrometry Sharon revealed that the proteasomes of archaea and a rabbit were also analyzed [Citation12].
“Specifically, we applied ion mobility, SID, collision induced dissociation, collision induced unfolding profiles and top-down pseudo-MS3 experiments in order to study the structural properties of the ortholog proteasomes”, noted Sharon, speaking to BioTechniques at the conference [Citation13].
The results of this analysis were surprising, as the structural properties of each species' 20S proteasome did not align with the linear increase in size and stability that was expected to follow the evolutionary development of the proteasome by the research team.
“We saw that yeast was the most stable and biggest proteasome, more than mammalian complexes, and it could be that stability is not a feature that is advantageous, maybe the more flexible structures are those that facilitate functionality. Indeed, the number of intrinsically unstructured proteins that are identified is growing, so maybe flexibility in structure is a feature that assists function”, Sharon theorized.
Challenges in nMS
The power of nMS as a tool for structural biology is clear to see, but it is important to remain critical of the process and to regard all results through a slightly reserved lens. Many researchers working with nMS will note the frequent skepticism that they meet when attempting to convince researchers from other fields that the analytes that fly through the spectrometer are indeed in their ‘native’ state.
This skepticism is by no means unfounded. The structures analyzed are commonly found in solutions or are taken from the membrane of the cell. To convince a molecular biologist that these structures to remain intact when in the gas phase takes a great deal of evidence to accomplish.
Gabelica, for instance, noted that in “…fields like structural chemistry and biophysics, there is always this suspicion about nMS, because we are analyzing things in the gas phase and we want to infer information on what existed in the solution. In every community, you have to convince and prove that what you are claiming to deduce from MS is valid. In different communities, it's a different challenge. For example, among organic chemists and supramolecular chemists, a few are really convinced, but most will want to crystallize the structure and analyze it by NMR, because that is more conventional and thus better accepted by their peers.”
In her own study into G-quadruplex structure, Gabelica noted herself that there is an element of structural difference in the solution structure compared with the gas phase structure of G-quadruplexes, as identified by ion mobility spectroscopy [Citation10].
These differences do not spell the doom of nMS as a technique, nor provide cause to deride and dismiss the results obtained by the procedure. They are identified and considered aspects of using these methods that any conscientious researcher should certainly consider when drawing conclusions and constructing experiments.
What these limitations truly highlight is that, while the instrumentation and technique is continually improving, the development of nMS is not yet complete. Further understanding of the alterations acquired in the transition from solution to gas phase are required alongside an established method accounting for or minimizing them. nMS is currently a powerful tool for structural biology, but there is plenty of work to do yet.
References
- Aston F . A positive ray spectrograph. Lond. Edinb. Phil. Mag.38(228), 707–714 (1919).
- Gault J , RobinsonC. Cracking complexes to build models of protein assemblies. ACS Cent. Sci.5(8), 1310–1311 (2019).
- Seffernick J , HarveyS, WysockiV, LindertS. Predicting protein complex structure from surface-induced dissociation mass spectrometry data. ACS Cent. Sci.5(8), 1330–1341 (2019).
- Struwe W , RobinsonC. Relating glycoprotein structural heterogeneity to function – insights from native mass spectrometry. Curr. Opin. Struct. Biol. (2019).
- Rose R , DamocE, DenisovE, MakarovA, HeckA. High-sensitivity Orbitrap mass analysis of intact macromolecular assemblies. Nat. Methods9(11), 1084–1086 (2012).
- Wu D , StruweW, HarveyD, FergusonM, RobinsonC. N-glycan microheterogeneity regulates interactions of plasma proteins. Proc. Natl Acad. Sci. USA115(35), 8763–8768 (2018).
- Dulfer J , KadekA, KopickiJD, KrichelB, UetrechtC. Structural mass spectrometry goes viral. Adv. Virus Res.105, 189–238 (2019).
- Nyíri K , HarrisM, MatejkaJet al. HDX and native mass spectrometry reveals the different structural basis for interaction of the staphylococcal pathogenicity island repressor Stl with dimeric and trimeric phage dUTPases. Biomolecules9(9), 488 (2019).
- Meng Z , Simmons-WillisTA, LimbachP. The use of mass spectrometry in genomics. Biomol. Eng.21(1), 1–13 (2004).
- D'Atri V , GabelicaV. DNA and RNA telomeric G-quadruplexes: what topology features can be inferred from ion mobility mass spectrometry?Analyst144(20), 6074–6088 (2019).
- BioTechniques. Valérie Gabelica on native mass spectrometry for nucleic acids. www.biotechniques.com/interview/valerie-gabelica-on-native-mass-spectrometry-for-nucleic-acid/
- Ben-Nissan G , VimerS, TarnavskyM, SharonM. Structural mass spectrometry approaches to study the 20S proteasome. Methods Enzymol.619, 179–223 (2019).
- BioTechniques. Probing the proteasome. www.biotechniques.com/chemical-biology-bio-and-analytical-chemistry/probing-the-proteasome/

