
Abstract
The implication of MET alterations in solid tumors and the immune microenvironment remains elusive. Formalin-fixed, paraffin-embedded samples of 21 patients with solid tumors harboring MET alterations were used for immunohistochemical staining. Extracted RNA was analyzed with the NanoString nCounter human PanCancer immune profiling panel (NanoString Technologies, Inc., WA, USA). Patients were diagnosed with lung (n = 10), breast (n = 5), genitourinary (n = 3) or colorectal cancer (n = 3). Eleven had a MET missense mutation, four had an exon 14 splice site mutation and six had MET amplification. CD6, CCL19, CD40LG, XCR1, MAGEA1, ATM and CCL19 genes were significantly differentially expressed in MET-altered cancers. MET alterations may have a role in various solid tumors as potential therapeutic targets and combination therapy candidates with immune checkpoint inhibitors.
Lay abstract
MET is a receptor for growth signals that keeps cells alive and healthy. However, some tumors have changes in MET that allow for uncontrollable cell growth. Patients with MET-altered tumors may benefit from treatments targeting this gene, but eventually they become resistant to the treatments. Thus, there is a need to identify additional therapies for this patient population. The authors tested immune gene expression in tumors with MET alterations to determine if these patients would benefit from a new class of treatments called immunotherapies and found that patients with and without MET changes had differences in immune gene expression.
Cancer is the second leading cause of death in the United States and is responsible for more than 1 million deaths worldwide annually [Citation1]. The MET receptor tyrosine kinase and its ligand hepatocyte growth factor have been implicated as key players in tumor cell migration, proliferation and invasion in a variety of cancers [Citation2]. Oncogenic driving mutations in the MET gene were first identified in hereditary papillary renal carcinoma. Subsequent studies identified similar alterations in sporadic renal carcinoma, non-small-cell lung cancer (NSCLC), breast cancer, colorectal cancer, gastric cancer, head and neck squamous cell carcinoma and other solid tumors [Citation3–8]. The authors' group was the first to establish the oncogenic role of MET alterations in lung cancer through the identification of mutations in the MET juxtamembrane domain in NSCLC and in the Sema domain in small cell lung cancer [Citation9–12]. Interestingly, additional studies have identified recurrent MET exon 14 skipping mutations in NSCLC and subsequent studies have demonstrated clinical efficacy of small molecule MET inhibitors such as capmatinib, tepotinib, crizotinib or cabozantanib for treatment of NS patients with these MET exon 14 skipping mutations [Citation13–15].
Aberrant MET activity has been implicated in acquired tumor cell resistance to treatment with EGFR-targeted therapy and, more recently, resistance to immunotherapy [Citation16,Citation17]. The involvement of MET in multiple cancers demonstrates its oncogenic significance and role in therapy resistance. Therefore, the authors' study aims to understand the molecular and immune profile of MET aberrations in solid tumors. To examine the immune profile in MET-altered tumors, the authors leveraged the NanoString nCounter human PanCancer immune profiling panel (NanoString Technologies, Inc., WA, USA) to perform multiplex gene expression analysis on 770 genes, including common checkpoint inhibitors, CT antigens and genes covering both the adaptive and innate immune response. Previous studies have shown that the nCounter technique can accurately analyze mRNA from formalin-fixed, paraffin-embedded (FFPE) tissue, with results similar to fresh frozen tissue [Citation18,Citation19]. MET receptor tyrosine kinase alterations have previously been shown to correlate with expression of immunoregulatory molecules, including PD-L1, and may play a role in immune evasion [Citation20,Citation21]. Therefore, this study was conducted to investigate the expression of immune markers in solid tumors with MET alterations to identify markers that may be associated with tumorigenesis. The aim of the study was to identify co-stimulatory and co-inhibitory immune markers in FFPE-derived mRNA of various solid tumors.
Methods
Seventy solid tumor patients with MET alterations or amplifications were initially identified. Of those patients, 21 had tissue available for further analysis. Patients had next-generation sequencing performed by their primary oncologist between 2010 and 2018 to identify MET alterations in the clinic. Data were collected between 2016 and 2018 through retrospective chart review and stored in a secure database. The study was approved by the City of Hope (COH) institutional review board (IRB) in accordance with an assurance filed with and approved by the Department of Health and Human Services at COH under IRB 17121 and was conducted according to the Declaration of Helsinki. Data were de-identified and analyzed anonymously.
Tissue samples
Representative FFPE samples of each tumor type (ten lung cancer, five breast cancer, three genitourinary cancer and three colorectal cancer) were used for analysis. Matched-pair FFPE specimens without MET alterations (nine lung cancer, two breast cancer, four genitourinary cancer and four colorectal cancer) were utilized as the MET wild-type control. Patients underwent a commercially available, broad-based genomic testing panel that utilized next-generation sequencing (Foundation Medicine, Inc., MA, USA) to determine their MET status. Genomic profiling was performed prospectively at the request of the treating oncologist for the purpose of clinical decision-making and was later retrospectively reviewed for the purpose of this study. The tissues utilized were archival specimens already banked under the IRB-approved COH protocol for procurement, management and distribution of human biological materials and health information for research (IRB 07047). Access to tissue and clinical data associated with the patients was granted by the COH IRB under IRB 17121.
Immunohistochemical staining
Histological sections at a thickness of 5 μm were deparaffinized with xylene and rehydrated through an alcohol graded series (100% ethanol, 95% ethanol, 70% ethanol) to water. Antigens were retrieved, and endogenous peroxidase activity was quenched using 3% hydrogen peroxide. After blocking, the sections were incubated with the primary antibodies human MET (Invitrogen, CA, USA), p-MET/Tyr1230/1234/1235 (Invitrogen, CA, USA), CD4 (LifeSpan BioSciences, Inc., WA, USA) and PD-L1 (LifeSpan BioSciences, Inc., WA, USA) and incubated for 30 min or 1 h at room temperature or at 4°C overnight depending on the optimization result of each antibody. Specimens were then incubated with the EnVision+ system horseradish peroxidase-labeled polymer, anti-mouse or anti-rabbit, correspondingly, for 30 min at room temperature, followed by incubation with the liquid DAB+ substrate chromogen system (Dako, CA, USA) for 8 min at room temperature on a Dako autostainer. After washing, the specimens were then counterstained with hematoxylin and covered with coverslips.
RNA extraction & gene expression profiling
Microdissection of FFPE samples was performed to enrich sample tumor purity. RNA was extracted from microdissected FFPE samples using the miRNeasy FFPE kit (Qiagen, MD, USA). RNA concentration was assessed using the NanoDrop ND-1000 spectrophotometer and Qubit 3.0 fluorometer (Thermo Fisher Scientific, MA, USA). RNA fragmentation and quality control were further determined using the 2100 Bioanalyzer (Agilent, CA, USA). FFPE RNA was applied to the NanoString nCounter human PanCancer immune profiling panel (category number XT-CSO-HIP1-12; NanoString Technologies, Inc.), a panel assessing expression of 770 genes. The NanoString nCounter profiling system (NanoString Technologies, Inc.) has been described previously [Citation22]. Briefly, samples were hybridized to probes for 16 h at 65°C. The post-hybridization probe–target mixture was then purified using the nCounter prep station and quantified with the nCounter digital analyzer (NanoString Technologies, Inc.), with 280 fields of view counted. Gene expression analysis was performed using nSolver 4.0 and NanoStringDiff 1.10.0 (NanoString Technologies, Inc.) [Citation19]. The nSolver tool (NanoString Technologies, Inc.) advanced analysis module uses the geNorm algorithm to input normalized gene expression using housekeeping genes and identifies differentially expressed genes using a negative binomial mixture model for lowly expressed genes and a simplified negative binomial model for highly expressed genes and adjusts for false discovery using the Benjamini–Yekutieli method. All genes passing a false discovery rate <0.05 were considered significant. The NanoStringDiff package (NanoString Technologies, Inc.) uses a negative binomial-based model appropriate for discrete count data and employs a normalization step incorporating data from the internal nCounter (NanoString Technologies, Inc.) positive and negative controls and the panel housekeeping controls to identify differentially expressed genes across groups. The package adjusts for false discovery using the Benjamini–Hochberg method. Genes passing the false discovery rate <0.05 cutoff were considered significant. The code for analysis is available at: https://github.com/jasminerethmeyer/MET.
Results
Data and specimen analyses were conducted on 21 patients with MET alterations. In the authors' study, a majority of patients were Caucasian (n = 13; 62%), male (n = 13; 62%), ≥60 years old (n = 12; 57%) and diagnosed with stage IV cancer (n = 19; 90%; ). Patients were diagnosed with lung cancer (n = 10), breast cancer (n = 5), genitourinary cancer (n = 3) or colorectal cancer (n = 3). Eleven (52%) patients had a MET missense mutation, four (19%) had an exon 14 splice site mutation and six (29%) had MET amplification. Of the 21 patients, four (19%) lung cancer patients were treated with targeted therapy specifically for MET mutations (e.g., crizotinib, glesatinib; ). In the three patients treated with crizotinib, two progressed within 6 months of treatment and one discontinued after 2 months because of severe adverse events. The patient treated with glesatinib continued treatment for 12 months. The remaining 17 patients were treated with various regimens, including chemotherapy, immunotherapy, aromatase inhibitors and CDK4/6 inhibitors.
Table 1. Patient characteristics.
Table 2. MET mutation therapies.
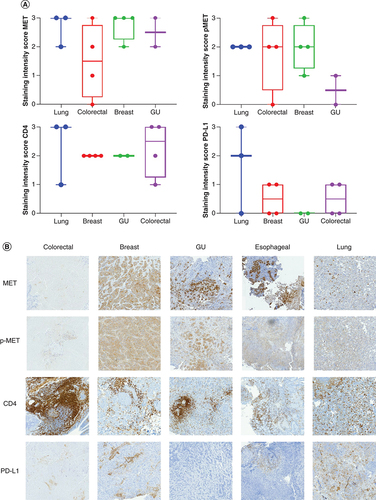
Patient specimens were collected, stained and scored for c-MET, p-MET, PD-L1 and CD4 (A & B). Overall, the staining intensity score for c-MET was relatively similar across three of the cancer types (lung, genitourinary and breast), with a slight decrease in intensity for colorectal cancer. The staining intensity score for p-MET was generally equal in lung, esophageal, colorectal and breast cancer cases, with genitourinary cancer cases showing little to no expression. The staining intensity score for PD-L1 was highest among the lung cancer cases and showed little to no expression in the remaining cancer types. The staining intensity score for CD4 was strongest in lung and colorectal cancer cases.
(A) Box plot representing the staining intensity IHC scores of c-MET, p-MET, PD-L1 and CD4 per disease type (lung, colorectal, breast and GU cancer). IHC staining of lung, colorectal, breast and GU tumor tissue with MET, p-MET, CD4 and PD-L1 antibodies was scored on a scale from 0 (no staining/no protein expression) to 3+ (strong staining/high protein expression). Staining intensity scores of MET, p-MET, CD4 and PD-L1 are mean ± SE. Statistical analyses were conducted using GraphPad Prism 8 by one-way ANOVA, followed by Tukey's multiple comparison test. No significant differences were seen between groups. (B) IHC staining of MET, p-MET, CD4 and PD-L1 for each disease type (colorectal, breast, GU and lung cancer).
ANOVA: Analysis of variance; GU: Genitourinary; IHC: Immunohistochemical; SE: Standard error.
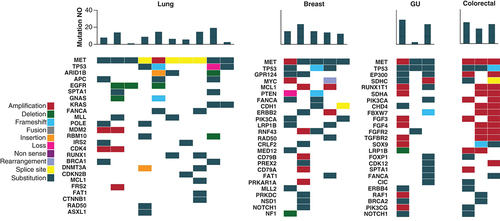
Molecular testing results from the electronic medical records of all 21 patients showed a number of co-mutations identified in each cancer type (). Overall, genitourinary (average 18.7 mutations) and colorectal (average 22 mutations) tumors had the highest number of total mutations compared with other disease types. The most frequent mutations in the lung cancer cohort were TP53 (n = 4; 40%), ARID1B (n = 3; 30%) and APC (n = 3; 30%). The most frequent mutations in the breast cancer cohort were TP53 (n = 3; 60%), GRP124 (n = 2; 40%) and MYC (n = 2; 40%). The most frequent mutations in the genitourinary cohort were TP53 (n = 3; 100%), EP300 (n = 2; 67%) and SDHC (n = 2; 67%). The most frequent mutations in the colorectal cancer cohort were TP53 (n = 3; 100%), BRCA2 (n = 2; 67%) and NOTCH3 (n = 2; 67%).
MET patients are categorized by cancer primary site. The heatmap demonstrates the genomic profile of each patient's tumor as well as the total number of mutations identified through NGS testing. The number of mutations per patient is shown in the bar chart at the top, with a heatmap of the genomic alterations identified underneath. The different types of genomic alterations identified in this cohort were classified as amplification, deletion, frameshift, fusion, insertion, loss, non-sense, rearrangement, splice site and substitution.
NGS: Next-generation sequencing; GU: Genitourinary; NO: Number.
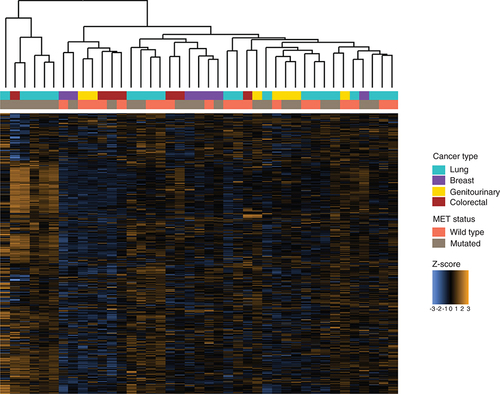
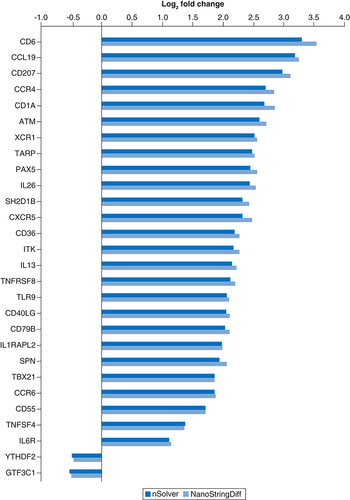
Gene expression profiling of patient tumor samples with and without MET alterations identified differences between these two groups. Hierarchical clustering of samples demonstrated a cluster of samples harboring MET mutations with elevated expression of genes from the multiplex assay of immune gene expression (). Thirty genes were identified as differentially expressed between samples with and without MET mutations using the nSolver tool (NanoString Technologies, Inc.; Supplementary Table 1), and 169 were identified as differentially expressed using NanoStringDiff (NanoString Technologies, Inc.; Supplementary Table 2), with 28 genes identified by both tools () [Citation19]. CD6, CCL19, ATM, CD40LG and XCR1 were identified as the most significantly differentially expressed by nSolver (NanoString Technologies, Inc.), and MAGEA1, ATM and CCL19 were identified by NanoStringDiff (NanoString Technologies, Inc.) as the most significant differentially expressed genes in MET-altered cancers. Interestingly, most differentially expressed genes were identified as upregulated in MET-mutated cancers compared with MET wild-type cancers.
Z-scores for 770 genes assayed using the multiplex gene expression panel in each tumor sample, annotated with cancer type and MET status.
All genes passing the FDR cutoff p < 0.05 for both tools are plotted. See Supplementary Tables for full lists of genes identified by each individual tool.
FDR: False discovery rate.
Discussion
In this study, the authors identified differential immune gene expression between patients with MET-altered cancers and those with MET wild-type cancers. Differential immune gene expression in patients with MET-altered tumors suggests these patients may benefit from treatment with immune-targeting therapies. Notably, the authors' study entailed a small sample size of patients with different cancer types, and additional work is needed to evaluate expression of these immune genes in a larger prospective cohort. Additionally, the authors' study examined gene expression from biopsies taken at diagnosis. Immune gene expression may fluctuate through the course of a patient's treatment. Thus, a longitudinal study of immune gene expression in MET-mutated cancers could be performed in the future to better inform the potential of immune-targeting therapies in this population; however, access to longitudinal samples from solid tumors for research purposes is limited.
Identification of differential gene expression in MET-altered tumors is important because aberrant MET activity has been observed in a wide variety of solid tumors, including NSCLC and breast and ovarian cancer [Citation23]. MET alterations in primary tumors commonly entail missense mutations or exon 14 skipping mutations, whereas in colorectal and NSCLC patients, MET amplification has been identified as a mechanism of acquired resistance following treatment with EGFR tyrosine kinase inhibitors [Citation24,Citation25]. MET exon 14 skipping mutations, which the authors have identified in both NSCLC and SCLC patients, cause deletion of the juxtamembrane domain containing the casitas B-lineage lymphoma E3-ubiquitin ligase binding site, leading to a decrease in the turnover of the resulting aberrant MET protein and prolonged signal activation [Citation6,Citation26]. Several studies have shown that small-molecule inhibitors of MET, such as crizotinib, cabozantinib, capmatinib and tepotinib, are effective in treating NSCLC patients with a MET exon 14 skipping mutation [Citation13]. For example, the phase II GEOMETRY mono-1 trial of capmatinib demonstrated increased overall response rates and improved median progression-free survival of 5.42 months in the cohort of patients receiving prior treatment and 9.69 months in the treatment-naive cohort [Citation14]. These results demonstrate the importance of identifying MET alterations early at initial diagnosis to improve patient response and survival through treatment with targeted therapy. Furthermore, a phase II trial of tepotinib has recently shown favorable results with a durable duration of response in MET-altered tumors [Citation15]. Capmatinib has been approved by the US FDA, and tepotinib has been approved in Japan for use in this group of patients. Patients have also been shown to derive improved benefit from MET-directed targeted therapies in various solid tumors that harbor MET amplifications or increases in MET gene copy number [Citation27]. Identifying the mechanisms for MET dependence of various solid tumors may be the key to developing novel MET therapeutics [Citation28].
Multiple studies have demonstrated a link between immune phenotypes and MET expression. For example, expression of MET has been implicated in activating myeloid-derived suppressor cells to suppress the immune system by hepatocyte growth factor secretion in mesenchymal stem cells [Citation29]. MET overexpression in breast cancer-associated adipose tissue composed of mesenchymal stem cells has also been correlated with recurrence and a potential role in tumorigenesis [Citation30]. In addition, a number of studies have recently shown that MET activation and amplification directly correlate with PD-L1 expression, and treatment of EGFR-resistant cells with a MET inhibitor has been shown to decrease PD-L1 protein and gene expression [Citation31–33]. IFN-γ has been revealed to induce PD-L1 expression that in turn limits the effectiveness of T-cell response [Citation34,Citation35], which may be actionable with MET inhibition. MET has been reported to restrain immunoresponse through transcriptional control of immunosuppressive molecules directly regulated by PD-L1 expression [Citation17,Citation31]; indeed, MET aberrations have been implicated in resistance to single-agent anti-PD-1/PD-L1 drugs [Citation36,Citation37]. Preclinical inhibition of MET signaling blocks PD-L1 upregulation, suggesting that MET inhibition may be an effective co-treatment alongside immune checkpoint inhibitors in patients with MET alterations [Citation17,Citation31]. Previous work has demonstrated that treating immune-competent mice with MET inhibitors complementary to immunotherapy leads to an increase in the number of active T cells and reduction of the proportion of exhausted T cells [Citation38]. Additionally, MET expression is able to activate CD4+ T cells and can induce tumor cell killing in natural killer/T-cell lymphoma cell lines, in which MET elicits an anti-tumor immune response by increasing the activation of T cells and reducing the synthesis of TGF-beta [Citation39]. Overall, this suggests that MET inhibition alongside immunotherapy inhibitors, such as anti-TGF-beta inhibitors, may potentiate immunotherapy efficacy, and not just in MET-driven tumors.
Identification of several differentially expressed chemokine receptors and CT antigens suggests a heterogeneous tumor immune microenvironment in MET-altered patients. In fact, chemokine receptors such as CCL19 and XCR1, alongside c-MET, have been shown to be overexpressed in mesenchymal stem cells [Citation40,Citation41]. CT antigens are tumor antigens that are typically expressed in germ cells of the testes as well as in a number of malignant tumors, but not in normal tissues [Citation42]. MAGEA1, a classic CT antigen, is a tumor-specific member of the MAGEA antigen family whose expression has been previously reported in several solid tumors [Citation43,Citation44]. Its expression has also been associated with progression and survival in NSCLC, but the specific mechanism for this phenomenon remains unknown [Citation45,Citation46]. CT antigens are ideal targets for cancer immunotherapy, and MAGEA1 has been shown to be actionable in vitro and in vivo by chimeric antigen receptor (CAR) T-cell therapy in lung adenocarcinoma cells, where the lung adenocarcinoma cells were effectively destroyed by IFN-γ secretion due to MAGEA1 inhibition [Citation47]. Thayaparan et al. reported that treatment of MET-expressing malignant mesothelioma cells with MET-retargeted CAR T cells was associated with significant IFN-γ release and tumor cell destruction [Citation48]. This suggests that co-expression of MAGEA1 and MET may have a role in immunoevasion and suppression of IFN-γ secretion [Citation49]. A clinical trial reported by Tchou et al. showed that CAR T-cell therapy was tolerated and evoked an inflammatory response within metastatic breast cancer patients [Citation50]. Several preclinical studies are currently considering MET a potential target for CAR T-cell therapy [Citation48,Citation51], and identification of co-stimulants of immune evasion, such as MAGEA1, may be important in optimizing therapy for solid tumors.
Conclusion and future perspective
Chemokine receptors and CT antigens could be utilized as co-stimulatory and co-inhibitory markers to identify patients with MET alterations who may benefit from immunotherapy or CAR T-cell therapy for several malignant solid tumors. The role of MET in a number of immune evasion mechanisms and upregulation of anti-cancer immunity receptors, such as PD-L1, must be clinically evaluated in combination therapy studies with immune checkpoint inhibitors. The nCounter technology (NanoString Technologies, Inc.) may serve as a reliable tool for assessing mRNA expression levels in FFPE tissue to determine clinically relevant immune markers associated with immune response or resistance.
MET mutations have been identified in a wide variety of solid tumors.
The efficacy of immunotherapies and the immune landscape of MET-altered tumors remain relatively unexplored.
The authors examined immune gene expression of patient tumor samples with and without MET alterations and identified changes in immune gene expression, including upregulation of CD6 and CD1A, in patients with MET-altered tumors.
Future studies are needed to examine the impact of immunotherapy treatment in the MET-altered patient population.
Author contributions
K L Reckamp: conceptualization, design, methodology, validation, formal analysis, original draft preparation, data accrual, tissue acquisition and writing review and editing. J McQuerry: conceptualization, design, methodology, validation, formal analysis, original draft preparation and writing review and editing. I Mambetsariev: conceptualization, design, methodology, data curation, original draft preparation and writing review and editing. R Pharaon: methodology, validation, data curation and writing review and editing. J Fricke: methodology, validation, data curation and writing review and editing. T Mirzapoiazova: methodology, validation, data curation and writing review and editing. R K Pillai: methodology, validation and tissue acquisition. Z Khan: validation, data accrual and tissue acquisition. M Fakih: validation, data accrual and tissue acquisition. Y Yuan: validation, data accrual and tissue acquisition. M Koczywas: validation, data accrual and tissue acquisition. E Massarelli: validation, data accrual and tissue acquisition. P Kulkarni: supervision, methodology and writing review and editing. S K Pal: supervision, methodology, data accrual, tissue acquisition and writing review and editing. M Sattler: methodology and writing review and editing. A Bild: supervision, conceptualization, design, methodology, validation, original draft preparation and writing review and editing. R Salgia: supervision, funding acquisition, conceptualization, design, methodology, validation, original draft preparation and writing review and editing. All authors contributed to the review, editing and approval of the final manuscript.
Ethical conduct of research
This study was approved by the City of Hope (COH) institutional review board (IRB) in accordance with an assurance filed with and approved by the Department of Health and Human Services at COH under IRB 17121 and was conducted according to the Declaration of Helsinki. Data were de-identified and analyzed anonymously. The tissues utilized were archival specimens already banked under the IRB-approved COH protocol for procurement, management and distribution of human biological materials and health information for research (IRB 07047). Access to tissue and clinical data associated with the patients was granted by the COH IRB under IRB 17121.
Supplemental Text 1
Download MS Word (30.2 KB)Supplemental Text 1
Download MS Word (30.2 KB)Acknowledgments
The authors thank City of Hope staff for their skill and dedication in helping the patients. The authors also thank Hongwei Holly Yin from the City of Hope molecular pathology core facility. Research reported in this publication includes work performed in the Pathology Research Services Core at City of Hope, supported by the National Cancer Institute of the NIH under award number P30CA033572. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at:www.tandfonline.com/doi/full/10.2217/epi-2016-0184
Financial & competing interests disclosure
This work was supported by the National Cancer Institute of the NIH under award numbers P30CA033572, U54CA209978, R01CA247471 and R01CA218545--and can be deposited on the NIHMS/PMC system. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
- SiegelRL , MillerKD , JemalA. Cancer statistics, 2020. CA Cancer J. Clin.70(1), 7–30 (2020).
- NakamuraT , NawaK , IchiharaA. Partial purification and characterization of hepatocyte growth factor from serum of hepatectomized rats. Biochem. Biophys. Res. Commun.122(3), 1450–1459 (1984).
- SchmidtL , DuhFM , ChenFet al.Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat. Genet.16(1), 68–73 (1997).
- DiRenzo MF , OliveroM , MartoneTet al.Somatic mutations of the MET oncogene are selected during metastatic spread of human HNSC carcinomas. Oncogene19(12), 1547–1555 (2000).
- GhadjarP , Blank-LissW , SimcockMet al.MET Y1253D-activating point mutation and development of distant metastasis in advanced head and neck cancers. Clin. Exp. Metastasis26(7), 809–815 (2009).
- deMelo Gagliato D , JardimDLF , FalchookGet al.Analysis of MET genetic aberrations in patients with breast cancer at MD Anderson phase I unit. Clin. Breast Cancer14(6), 468–474 (2014).
- NeklasonDW , DoneMW , SargentNRet al.Activating mutation in MET oncogene in familial colorectal cancer. BMC Cancer11, 424 (2011).
- SeiwertTY , JagadeeswaranR , FaoroLet al.The MET receptor tyrosine kinase is a potential novel therapeutic target for head and neck squamous cell carcinoma. Cancer Res.69(7), 3021–3031 (2009).
- MaPC , JagadeeswaranR , JagadeeshSet al.Functional expression and mutations of c-MET and its therapeutic inhibition with SU11274 and small interfering RNA in non-small cell lung cancer. Cancer Res.65(4), 1479–1488 (2005).
- MaPC , KijimaT , MaulikGet al.c-MET mutational analysis in small cell lung cancer: novel juxtamembrane domain mutations regulating cytoskeletal functions. Cancer Res.63(19), 6272–6281 (2003).
- SattlerM , SalgiaR. MET in the driver's seat: exon 14 skipping mutations as actionable targets in lung cancer. J. Thorac. Oncol.11(9), 1381–1383 (2016).
- MaulikG , KijimaT , MaPCet al.Modulation of the c-MET/hepatocyte growth factor pathway in small cell lung cancer. Clin. Cancer Res.8(2), 620–627 (2002).
- PaikPK , DrilonA , FanP-Det al.Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov.5(8), 842–849 (2015).
- WolfJ , SetoT , HanJ-Yet al.Capmatinib (INC280) in METΔex14-mutated advanced non-small cell lung cancer (NSCLC): efficacy data from the phase II GEOMETRY mono-1 study. J. Clin. Oncol.37(Suppl. 15), 9004–9004 (2019).
- PaikPK , FelipE , VeillonRet al.Tepotinib in non-small-cell lung cancer with MET exon 14 skipping mutations. 383(10), 931–943 (2020).
- CappuzzoF , JannePA , SkokanMet al.MET increased gene copy number and primary resistance to gefitinib therapy in non-small-cell lung cancer patients. Ann. Oncol.20(2), 298–304 (2009).
- MartinV , ChiriacoC , ModicaCet al.MET inhibition revokes IFNγ-induction of PD-1 ligands in MET-amplified tumours. Br. J. Cancer120(5), 527–536 (2019).
- ReisPP , WaldronL , GoswamiRSet al.mRNA transcript quantification in archival samples using multiplexed, color-coded probes. BMC Biotechnol.11, 46 (2011).
- WangH , HorbinskiC , WuHet al.NanoStringDiff: a novel statistical method for differential expression analysis based on NanoString nCounter data. Nucleic Acids Res.44(20), e151 (2016).
- TitmarshHF , O'ConnorR , DhaliwalK , AkramAR. The emerging role of the c-MET-HGF axis in non-small lung cancer tumor immunology and immunotherapy. Front. Oncol.10, 54 (2020).
- AhnHK , KimS , KwonDet al.MET receptor tyrosine kinase regulates the expression of co-stimulatory and co-inhibitory molecules in tumor cells and contributes to PD-L1-mediated suppression of immune cell function. Int. J. Mol. Sci.20(17), 4287 (2019).
- GeissGK , BumgarnerRE , BirdittBet al.Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat. Biotechnol.26(3), 317–325 (2008).
- MaPC , TretiakovaMS , MacKinnonACet al.Expression and mutational analysis of MET in human solid cancers. Genes Chromosomes Cancer47(12), 1025–1037 (2008).
- EngelmanJA , ZejnullahuK , MitsudomiTet al.MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science316(5827), 1039–1043 (2007).
- RaghavK , MorrisV , TangCet al.MET amplification in metastatic colorectal cancer: an acquired response to EGFR inhibition, not a de novo phenomenon. Oncotarget7(34), 54627–54631 (2016).
- TanYH , KrishnaswamyS , NandiSet al.CBL is frequently altered in lung cancers: its relationship to mutations in MET and EGFR tyrosine kinases. PLoS ONE5(1), e8972 (2010).
- GuoR , LuoJ , ChangJ , RekhtmanN , ArcilaM , DrilonA. MET-dependent solid tumours–molecular diagnosis and targeted therapy. Nat. Rev. Clin. Oncol.17(9), 569–587 (2020).
- SalgiaR , SattlerM , ScheeleJ , StrohC , FelipE. The promise of selective MET inhibitors in non-small cell lung cancer with MET exon 14 skipping. Cancer Treat. Rev.87, 102022 (2020).
- YenBL , YenML , HsuPJet al.Multipotent human mesenchymal stromal cells mediate expansion of myeloid-derived suppressor cells via hepatocyte growth factor/c-MET and STAT3. Stem Cell Reports1(2), 139–151 (2013).
- EternoV , ZambelliA , PavesiLet al.Adipose-derived mesenchymal stem cells (ASCs) may favour breast cancer recurrence via HGF/c-MET signaling. Oncotarget5(3), 613–633 (2014).
- SaigiM , Alburquerque-BejarJJ , McLeer-Florin Aet al.MET-oncogenic and JAK2-inactivating alterations are independent factors that affect regulation of PD-L1 expression in lung cancer. Clin. Cancer Res.24(18), 4579–4587 (2018).
- AlbitarM , SudarsanamS , MaWet al.Correlation of MET gene amplification and TP53 mutation with PD-L1 expression in non-small cell lung cancer. Oncotarget9(17), 13682–13693 (2018).
- DemuthC , AndersenMN , JakobsenKR , MadsenAT , SørensenBS. Increased PD-L1 expression in erlotinib-resistant NSCLC cells with MET gene amplification is reversed upon MET-TKI treatment. Oncotarget8(40), 68221–68229 (2017).
- BlankC , BrownI , PetersonACet al.PD-L1/B7H-1 inhibits the effector phase of tumor rejection by T cell receptor (TCR) transgenic CD8+ T cells. Cancer Res.64(3), 1140–1145 (2004).
- Garcia-DiazA , ShinDS , MorenoBHet al.Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep.19(6), 1189–1201 (2017).
- XingX , GuoJ , DingGet al.Analysis of PD1, PDL1, PDL2 expression and T cells infiltration in 1014 gastric cancer patients. Oncoimmunology7(3), e1356144 (2018).
- SabariJK , LeonardiGC , ShuCAet al.PD-L1 expression, tumor mutational burden, and response to immunotherapy in patients with MET exon 14 altered lung cancers. Ann. Oncol.29(10), 2085–2091 (2018).
- GloddeN , BaldT , vanden Boorn-Konijnenberg Det al.Reactive neutrophil responses dependent on the receptor tyrosine kinase c-MET limit cancer immunotherapy. Immunity47(4), 789–802.e789 (2017).
- KumaiT , MatsudaY , OhkuriTet al.c-Met is a novel tumor associated antigen for T-cell based immunotherapy against NK/T cell lymphoma. Oncoimmunology4(2), e976077 (2015).
- SpaethE , KloppA , DembinskiJ , AndreeffM , MariniF. Inflammation and tumor microenvironments: defining the migratory itinerary of mesenchymal stem cells. Gene Ther.15(10), 730–738 (2008).
- SonBR , Marquez-CurtisLA , KuciaMet al.Migration of bone marrow and cord blood mesenchymal stem cells in vitro is regulated by stromal-derived factor-1-CXCR4 and hepatocyte growth factor-c-MET axes and involves matrix metalloproteinases. Stem Cells24(5), 1254–1264 (2006).
- SimpsonAJ , CaballeroOL , JungbluthA , ChenYT , OldLJ. Cancer/testis antigens, gametogenesis and cancer. Nat. Rev. Cancer5(8), 615–625 (2005).
- ChomezP , DeBacker O , BertrandM , DePlaen E , BoonT , LucasS. An overview of the MAGE gene family with the identification of all human members of the family. Cancer Res.61(14), 5544–5551 (2001).
- NohST , LeeHS , LimSJet al.MAGE-A1-6 expression in patients with head and neck squamous cell carcinoma: impact on clinical patterns and oncologic outcomes. Int. J. Clin. Oncol.21(5), 875–882 (2016).
- YiE , ChangJE , LeemC , JeonCH , JheonS. Association of MAGE A1-6 expression with lung cancer progression. J. Cancer8(8), 1324–1329 (2017).
- MecklenburgI , SienelW , SchmidS , PasslickB , KuferP. A threshold of systemic MAGE-A gene expression predicting survival in resected non-small cell lung cancer. Clin. Cancer Res.23(5), 1213–1219 (2017).
- MaoY , FanW , HuHet al.MAGE-A1 in lung adenocarcinoma as a promising target of chimeric antigen receptor T cells. J. Hematol. Oncol.12(1), 106 (2019).
- ThayaparanT , PetrovicRM , AchkovaDYet al.CAR T-cell immunotherapy of MET-expressing malignant mesothelioma. Oncoimmunology6(12), e1363137–e1363137 (2017).
- MojicM , TakedaK , HayakawaY. The dark side of IFN-γ: its role in promoting cancer immunoevasion. Int. J. Mol. Sci.19(1), 89 (2017).
- TchouJ , ZhaoY , LevineBLet al.Safety and efficacy of intratumoral injections of chimeric antigen receptor (CAR) T cells in metastatic breast cancer. Cancer Immunol. Res.5(12), 1152–1161 (2017).
- LiuB , LiuZZ , ZhouMLet al.Development of c-MET-specific chimeric antigen receptor-engineered natural killer cells with cytotoxic effects on human liver cancer HepG2 cells. Mol. Med. Rep.20(3), 2823–2831 (2019).