Abstract
Viral neuraminidase inhibitors such as oseltamivir and zanamivir prevent early virus multiplication by blocking sialic acid cleavage on host cells. These drugs are effective for the treatment of a variety of influenza subtypes, including swine flu (H1N1). The binding site for these drugs is well established and they were designed based on computational docking studies. We show here that some common natural products have moderate inhibitory activity for H1N1 neuraminidase under docking studies. Significantly, docking studies using AutoDock for biligand and triligand forms of these compounds (camphor, menthol, and methyl salicylate linked via methylene bridges) indicate that they may bind in combination with high affinity to the H1N1 neuraminidase active site. These results also indicate that chemically linked biligands and triligands of these natural products could provide a new class of drug leads for the prevention and treatment of influenza. This study also highlights the need for a multiligand docking algorithm to understand better the mode of action of natural products, wherein multiple active ingredients are present.
Introduction
Hemagglutinin and neuraminidase are the two key glycoproteins responsible for viral influenza infection.Citation1 Hemagglutinin is present on the surface of the virion and is needed for infection, while neuraminidase is responsible for cleavage of sialic acid (neuraminic acid) from glycans of the infected cell.Citation2,Citation3 These two proteins are drug targets for viral infections, and the neuraminidase inhibitors, oseltamivir (Tamiflu®, Roche, Basel, Switzerland) and zanamivir (Relenza®, Philadelphia, PA, USA), are broad spectrum antiviral drugs, useful for the treatment of a variety of forms of influenza.Citation4,Citation5 The World Health Organization recommends the use of oseltamivir or zanamivir for the treatment of H1N1 virus, and patient recovery with these drugs has been impressive. Oseltamivir and zanamivir are reversible competitive inhibitors of neuraminidase, thereby preventing virion release from infected cells.Citation6 The binding site interaction of these drugs is well established, and they are amongst only a few drugs with binding sites based on computational docking and quantitative structure-activity relationships that are well understood.
The success of neuraminidase inhibitors can be attributed to the sequence and structure conservation of neuraminidase in all subtypes of influenza. Reports of mutations in neuraminidase are relatively sparse compared with other viral protein domains.Citation1 There are significant changes in both hemagglutinin and neuraminidase protein of the 2012 H1N1 virus, ie, 27.2% and 18.2% of the amino acid sequence, respectively, from 2008 H1N1 isolates.Citation1 Though these changes are a complication with respect to the use of existing influenza vaccines, the positive response of this virus to current neuraminidase inhibitors indicates that this change does not significantly alter the protein-ligand interactions.
Hence, neuraminidase is an excellent general target for the control of viral influenza, including H1N1. Although neuraminidase inhibitors such as oseltamivir and zanamivir are approved for both treatment and prevention of influenza, more effective preventive medications would be useful in slowing the spread of H1N1 influenza.Citation4,Citation7 Therefore, there is a need to develop improved drugs which can prevent the spread of the virus.
There are several common cold medications which have been available for generations to provide relief from symptoms, including sneezing, blocked nose, sore throat, and nasal congestion. They include over-the-counter products (such as inhalers and vaporizers) and homemade decoctions of spices (eg, ginger, pepper, basil). Most of these have a pungent aromatic odor and the exact mechanism of action of their active constituents is unknown. The effectiveness of these compounds in relieving symptoms caused by influenza warrants further investigation into whether any of this efficacy comes from specific binding mechanisms. If these compounds selectively bind to specific targets then they could potentially be used more broadly in H1N1 influenza prevention or treatment.
Materials and methods
The Protein Data Bank crystal structure (2hu0) of the neuraminidase in H5N1 bound to oseltamivir was used as a referenceCitation8 and a model built for H1N1 neuraminidase was used for docking studies.Citation9 AutoDockCitation10 (The Scripps Research Institute, La Jolla, CA, USA) was used for protein-ligand docking studies. Chemical structures were downloaded from Pubchem and similar databases and verified or drawn using ACD Chemsketch (Advanced Chemistry Development, Inc., Toronto, ON, Canada) Protein electrostatic potential was calculated using the Adaptive Poisson Boltzmann Solver (ABPS) with AutoDockTools release 1.5.4.Citation10 Molecular graphics images were produced using the UCSF Chimera package from the Computer Graphics Laboratory, University of California, San Francisco, CA, USA.Citation11
Results and discussion
Neuraminidase structure selection and protein model
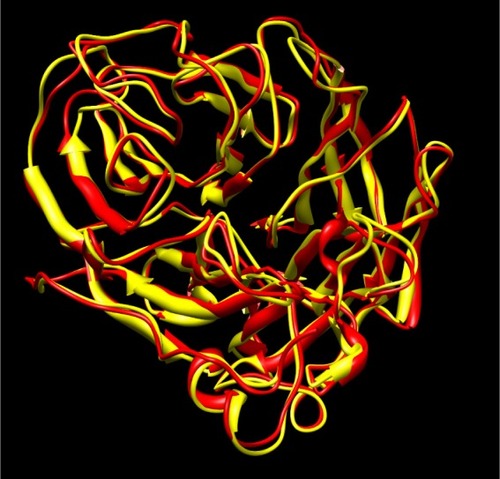
Maurer-Stroh et al have demonstrated that the neuraminidase present in current variants of H1N1 is similar to that present in H5N1 avian influenza.Citation9 The crystal structure of neuraminidase in H5N1 bound to oseltamivir is known and we used this as a reference structure for our study. MaurerStroh et al built a model for H1N1 based on the previously reported information for H5N1, and we used the same model after ensuring that the crystal structure of neuraminidase in H5N1 and H1N1 is similar (root-mean-square deviation 0.81 Å). shows the overlay of H5N1 and H1N1 neuraminidase structures. Recent crystal structures of some strains of H1N1 neuraminidase, such as the 1918 influenza viral strain, indicate that there is a high degree of structural similarity to the H5N1 strain. Both are group 1 subtypes characterized by a similar conformation for the 150 loop or 150 cavity which, on binding to zanamivir, undergoes conformational change that is similar for both H1N1 and H5N1 subtypes.Citation12
Figure 1 Overlay of H5N1 crystal structure (2hu0, yellow) and the modeled structure of H1N1 (red). The root-mean-square deviation of the two structures is 0.81 Å.
Binding site of H1N1 neuraminidase
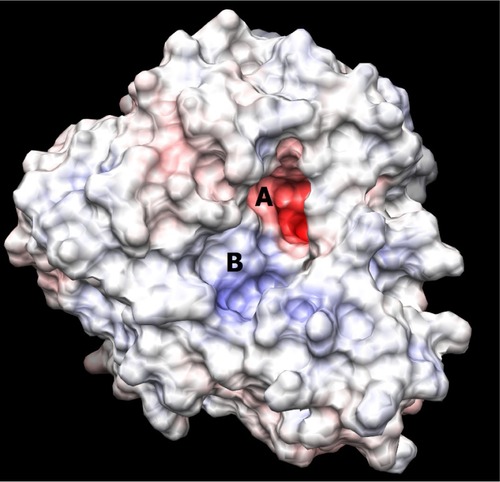
The binding site of neuraminidase is a conserved region in different variants of influenza A and B. The key step of viral attack and multiplication in the host cell is driven by binding of viral neuraminidase to sialic acid on the human cell surface.Citation3 Hence, the inhibitors of neuraminidase (oseltamivir and zanamivir) are designed in such a way that they are chemically as similar as possible to sialic acid, with improved enzyme affinity. shows the binding site of H1N1 neuraminidase, depicting the two different electrostatic grooves as A and B, representing two complementary regions, a highly negatively charged region (depicted by red color), and a positively charged stretch. Under docking conditions, sialic acid has the possibility of nine hydrogen bonds at the A binding site and synthetic inhibitors like oseltamivir and zanamivir have about seven. Important aspects for the H1N1 neuraminidase quantitative structure-activity relationship are the electrostatic interaction between ligand and protein, and the possible formation of salt bridges between the amine-rich terminus of the ligand and glutamic acid in the binding site. This kind of ligand design is dependent on interaction between the binding site amino acid residues and the ligand, and is probably a reason why oseltamivir-resistant strains have started to evolve.Citation13–Citation15
Figure 2 Picture depicting the two regions of the H1N1 neuraminidase binding site. The red region (A) is the electronegative region, where the amine-rich functional groups of ligands bind, and the large blue region (B) a large electropositive zone. The deep binding pocket (A) could host many kinds of ligands, but high affinity can be obtained only when there is a well defined functional group interaction (electrostatic and/or structural).
Binding of oseltamivir and sialic acid to H1N1 neuraminidase
Oseltamivir is one of the few inhibitors of neuraminidase with subnanomolar affinity and currently in clinical use for the treatment of influenza.Citation16 In our docking mode, oseltamivir is the strongest ligand of those studied (), with the lowest docking energy of −13.94 kcal/mol. The predicted binding constant for oseltamivir is at least 10 times higher than that for sialic acid (). The hydrogen bond-rich interaction of sialic acid has not resulted in higher binding affinity. The higher affinity of oseltamivir is probably related to salt bridge formation (). In our analysis, zanamivir had a binding affinity similar to that of sialic acid, with docking energy of −12.84 kcal/mol ().
Figure 3 Model showing docked configuration of sialic acid with H1N1 neuraminidase.
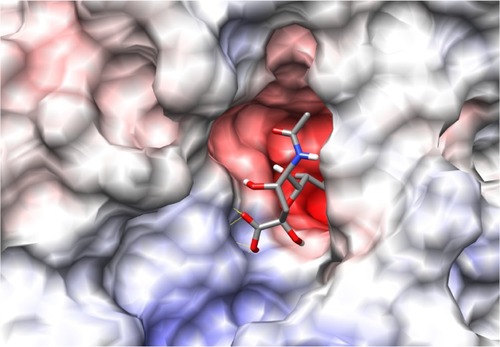
Figure 4 Model showing docked configuration of oseltamivir with H1N1 neuraminidase.
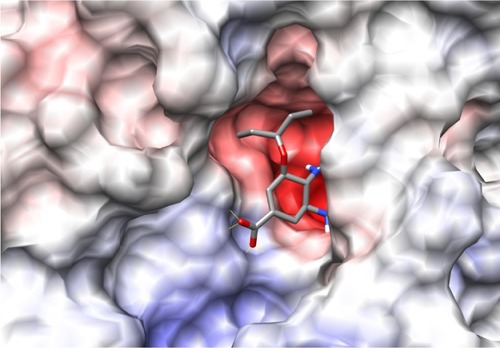
Table 1 Docking energies (affinity) for different chemicals with H1N1 neuraminidase
Zanamivir also forms a binding site salt bridge and has seven hydrogen bonds possible at the binding site. The significantly higher binding affinity of oseltamivir as compared with zanamivir, even though both have a similar number of hydrogen bonds and form salt bridges, indicates that there may be a structural parameter that impacts binding that was not determined in previous studies.Citation14,Citation15,Citation17
Docking of natural product chemical constituents
We docked the three major chemical constituents of common inhalers, namely menthol, camphor, and methyl salicylate. These components as well as antioxidants (rosemary oil components), traditional cold medicines (ginger, pepper) and common natural essential oil products have shown significant affinity for H1N1 neuraminidase (). Surprisingly, we observed that a steroid molecule (oleanolic acid) present in rosemary extract has high affinity for H1N1 neuraminidase (−11.74 kcal/mol). Oleanolic acid does not bind with as many hydrogen bonds as sialic acid and oseltamivir, indicating that it has a structural specificity for the neuraminidase binding site. Cold and influenza relief recipes used in traditional Indian medicine are general decoctions of herbs and often use cocktails containing multiple ingredients.Citation18,Citation19 These would have multiple active components, which may have cumulative or synergistic actions. Based on this, we evaluated the binding of some of these compounds in combination. Since the docking software accepts only one molecule at a time, we evaluated the combination effect using a “linked-structure” approach. As seen in , these combination linked structures had significantly higher binding affinities than the individual molecules.
Multiligand binding: linked structure approach
Of the natural products evaluated, the most potent H1N1 neuraminidase binders in our model were oleanolic acid and rosmarinic acid. It is possible that multiple components could simultaneously bind to the of H1N1 neuraminidase. AutoDock does not allow multiple ligands to be docked with the same protein at the same time. To the best of our knowledge, there is no docking program which allows multiple ligand docking. Simultaneous binding of ligands to the same target is an unusual concept, but it has been used in biligand design for some protein targets, such as oxidoreductases.Citation20 This approach uses docking and nuclear magnetic resonance studies for two compounds binding next to each other, using NOE determined distances to show the spatial relationship between each compound. A new compound containing both molecular structures can then be synthesized and tested for binding affinity. This biligand approach has resulted in some clinical leads and the linking of two active compounds through methylene groups has shown some success. In the case of H1N1 neuraminidase, the binding site is so large that it can readily accommodate more than one ligand. Neuraminidase is similar to oxidoreductase in that both have two binding sites A and B adjacent to each other.Citation21
We hypothesized that if a ligand was structurally connected through a methylene group at the methyl terminus of a molecule it should provide a dockable biligand with sufficient flexibility due to two additional single bonds, which could mimic the simultaneous binding of two ligands. Among the functional groups commonly present in both natural and synthetic inhibitors, methyl groups are physiologically the least relevant due to their small size and lack of chemical functionality. Methylene linking has been successfully used in biligand development for oxidoreductases and binding shown to occur using nuclear magnetic resonance. The methylene group itself has minimal impact on binding and the methylenes primarily enable flexibility in orientation of the biligands. Hence, conjugation to a terminal methyl group may not significantly change the affinity of the original ligand. Secondly, when two molecules are chemically linked, the degrees of freedom for rotation decreases, which could affect the docking results, in that docking software may not be able to test all possible orientations. By bridging methyl groups of ligands with a methylene group, we created two extra rotatable bonds, which compensates for the loss of degrees of freedom. Finally, introduction of an extra methylene functional group creates an average 3 Å distance between molecules, which is the minimum distance two molecules are expected to maintain when they are bound together in solid state to a target.
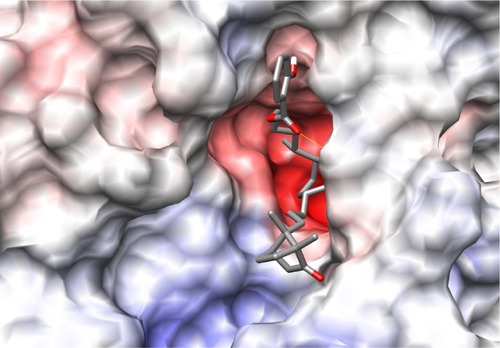
The first combination ligand we designed was camphor-menthol. This molecule is predicted to bind with a docking energy of −9.31 kcal/mol, which is about 1000-fold higher than the affinity of each individual chemical component. This combination molecule formed only one hydrogen bond, indicating that structural affinity rather than electrostatic interaction is primarily responsible for the increased binding affinity. We then designed a triligand comprising menthol, camphor, and methyl salicylate, with three possible structural forms. We followed the methylene linking strategy in designing these molecules, and they were methyl salicylate-menthol-camphor, methyl salicylate-camphor-menthol, and camphor-methyl salicylate-menthol (). We observed that all three combination ligands had good affinity for H1N1 neuraminidase, but the mode and orientation of binding was different. Among these, methyl salicylate-menthol-camphor had the highest affinity, which was almost the same as sialic acid and zanamivir (). The binding of methyl salicylate-menthol-camphor resembles a triangular structure covering the electronegative pocket in the binding region (). Importantly, the methyl salicylate-menthol-camphor is bound completely at the A site and no part of the molecule was interacting with the B site. This indicates that the interaction of this ligand with H1N1 neuraminidase has a strong structural basis.
Figure 5 Chemical structure of combination ligands. Methyl salicylate-menthol-camphor (A), methyl salicylate-camphor-menthol (B), and camphor-methyl salicylate-menthol (C). The arrows show the methylene bridges artificially created to enable the possibility of docking the combination ligand with the enzyme. Note the drop in docking energy of B (−11.18 kcal/mol) when menthol and camphor position is interchanged from A (−12.68).
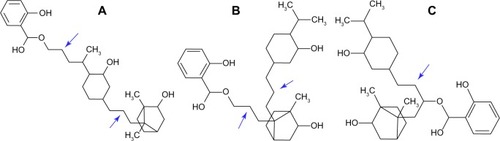
Figure 6 Docking of methyl salicylate-menthol-camphor to neuraminidase enzyme.
In the case of methyl salicylate-camphor-menthol (), the menthol group occupied the B site. This is in contrast with methyl salicylate-menthol-camphor, where there was a specific stabilizing interaction with the A binding site (). This specific interaction was absent with methyl salicylate-camphor-menthol and may be responsible for the lower docking energy of this triligand (−11.18 versus −12.68 kcal/mol, refer to ).
Figure 7 Docking of methyl salicylate-camphor-menthol with neuraminidase.
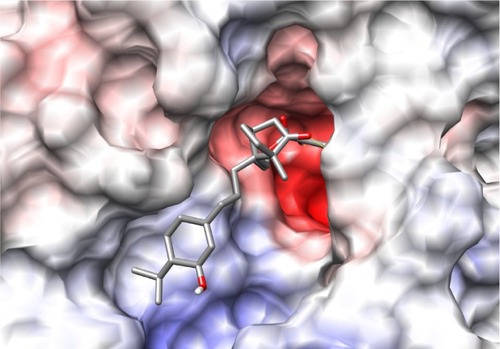
The difference in affinity of other combination ligands with respect to methyl salicylate-menthol-camphor suggests that there could be specific structural sites for the three ligands, and interchanging their position in the binding site alters the overall binding affinity.
Simultaneous reversible binding of multiple ligands
Docking studies are a good initial method of analysis that can provide binding information for these complex natural products. These ligands can bind reversibly to H1N1 neuraminidase, with affinity similar to sialic acid, indicating that H1N1 neuraminidase attack on host cells is likely to be inhibited. Current docking algorithms do not allow multiple-ligand simultaneous docking to target protein. Most natural products are known to have multiple active components, providing cumulative or synergistic effects, also overcoming buildup of resistance. It is highly desirable to have new molecular docking software capable of multiligand docking, for analyzing and understanding the mode of action of natural products.
Possible experimental verification of the combination ligand hypothesis
The neuraminidase enzyme is a well studied system, and protocols for its expression, purification, and crystallization have been reported in the literature.Citation22 The binding of multiple ligands could be verified by crystallizing neuraminidase in the presence of varying ratios of these constituents, where difficulties in crystallization due to high volatility would need to be overcome. Synthesis of the larger and probably less volatile biligands and triligands and their cocrystallization with H1N1 neuraminidase could also be tested experimentally. It is likely that some of the increased binding energy observed for biligands and triligands is due to increased ligand size, resulting in an increased hydrophobic contact surface area, so synthesis and experimental verification of H1N1 neuraminidase binding is particularly important to validate the potential of these ligands as drug leads. Alternatively, clinical evaluation would give insight into the benefits of these types of potential medications. Given that compounds like oleanolic acid, rosmarinic acid, methyl salicylate, menthol, and camphor are commonly used ingredients with low toxicity, there would be minimal concern regarding safety and adverse effects during a clinical study. It is possible that observation and population studies would provide useful initial clinical information on any protective effect of these compounds.
Conclusion
Modeling studies of biligands and triligands, created by linking methyl salicylate, menthol, and camphor via methylene bridges, indicate that in combination these compounds may be potential inhibitors of H1N1 neuraminidase. It is important to note, however, that high docking energies are indicative but not necessarily predictive of binding affinity. Therefore, these compounds need to be synthesized and tested experimentally to determine if they in fact have high binding affinity for H1N1 neuraminidase.
We have shown that the strong hydrogen bond-driven affinity of zanamivir, sialic acid, and oseltamivir is not the mechanism for methyl salicylate-menthol-camphor binding. The hydrophobic and structural interaction of these natural products indicates sequential and reversible binding to H1N1 neuraminidase. The structural and hydrophobic interactions of these chemical constituents offer the possibility of activity against H1N1 neuraminidase. By designing biligands and triligands, we were able to use AutoDock to study multiligand binding. A combination of such small molecules or these biligands and triligands may offer a new class of lead drug candidates for the prevention or treatment of influenza.
Acknowledgments
This research was supported by a grant from the Indian Council of Medical Research.
Disclosure
The authors report no conflicts of interest in this work.
References
- GallaherWRTowards a sane and rational approach to management of influenza H1N1Virol J20096515719422701
- HarrisonSCViral membrane fusionNat Struct Mol Biol20081569069818596815
- BurmeisterWPRuigrokRWHCusackSThe 2.2 A resolution crystal structure of influenza B neuraminidase and its complex with sialic acidEMBO J19921149561740114
- HeinonenSSilvennoinenHLehtinenPEarly oseltamivir treatment of influenza in children 1–3 years of age: a randomized controlled trialClin Infect Dis20105188789420815736
- CrusatMde JongMDNeuraminidase inhibitors and their role in avian and pandemic influenzaAntivir Ther20071259360217944267
- De ClercqEAntiviral agents active against influenza A virusesNat Rev Drug Discov200651015102317139286
- EilandLSEliandEHZanamivir for the prevention of influenza in adults and children age 5 years and olderTher Clin Risk Manag2007346146518488077
- RussellRJHaireLFStevensDJThe structure of H5N1 avian influenza neuraminidase suggests new opportunities for drug designNature2006443454916915235
- Maurer-StrohSMaJLeeRTSirotaFLEisenhaberFMapping the sequence mutations of the 2009 H1N1 influenza A virus neuraminidase relative to drug and antibody binding sitesBiol Direct200941919144117
- MorrisGMHueyRLindstromWAutodock4 and AutoDockTools4: automated docking with selective receptor flexibilityJ Comput Chem2009302785279119399780
- PettersenEFGoddardTDHuangCCUCSF chimera – a visualization system for exploratory research and analysisJ Comput Chem2004251605161215264254
- XuXZhuXDwekRAStevensJWilsonIAStructural characterization of the 1918 Influenza Virus H1N1 neuraminidaseJ Virol200882104931050118715929
- WangNXZhengJJComputational studies of H5N1 influenza virus resistance to oseltamivirProtein Sci20091870771519309695
- CollinsPJHaireLFLinYPCrystal structures of oseltamivir-resistant influenza virus neuraminidase mutantsNature20084531258126118480754
- LinLLiYZhangLHouTTheoretical studies on the susceptibility of oseltamivir against variants of 2009 A/H1N1 influenza neuraminidaseJ Chem Inf Model2012522715272922998323
- SchirmerPHolodniyMOseltamivir for treatment and prophylaxis of influenza infectionExpert Opin Drug Saf2009835737119355841
- HanNLiuXMuYExploring the mechanism of zanamivir resistance in a neuraminidase mutant: a molecular dynamics studyPLoS One20127e4405722970161
- LeeSJUmanoKShibamotoTLeeKGIdentification of volatile components in basil (Ocimum basilicum L.) and their antioxidant propertiesFood Chem200591131137
- BallabhBChaurasiaOPTraditional medicinal plants of cold desert Ladakh: used in treatment of cold, cough and feverJ Ethnopharmacol200711234134917459623
- SemDSBertolaetBBakerBSystems-based design of bi-ligand inhibitors of oxidoreductases: filling the chemical proteomic toolboxChem Biol20041118519415123280
- ColmanPMInfluenza virus neuraminidase: structure, antibodies, and inhibitorsProtein Sci19943168716967849585
- SchmidtPMAttwoodRMMohrPGBarrettSAMcKimm-BreschkinJLA generic system for the expression and purification of soluble and stable influenza neuraminidasePLoS One20116113