
Abstract
Resistance to antiestrogen therapy remains a significant problem in breast cancer. Low expression of inhibitor of growth 4 (ING4) in primary tumors has been correlated with increased rates of recurrence in estrogen receptor-positive (ER+) breast cancer patients, suggesting a role for ING4 in ER signaling. This study provides evidence that ING4 inhibits ER activity. ING4 overexpression increased the sensitivity of T47D and MCF7 ER+ breast cancer cells to hormone deprivation. ING4 attenuated maximal estrogen-dependent cell growth without affecting the dose–response of estrogen. These results indicated that ING4 functions as a noncompetitive inhibitor of estrogen signaling and may inhibit estrogen-independent ER activity. Supportive of this, treatment with fulvestrant but not tamoxifen rendered T47D cells sensitive to hormone deprivation as did ING4 overexpression. ING4 did not affect nuclear ERα protein expression, but repressed selective ER-target gene transcription. Taken together, these results demonstrated that ING4 inhibited estrogen-independent ER activity, suggesting that ING4-low breast tumors recur faster due to estrogen-independent ER activity that renders tamoxifen less effective. This study puts forth fulvestrant as a proposed therapy choice for patients with ING4-low ER+ breast tumors.
Introduction
Estrogen receptor (ER) signaling plays a pivotal role in breast cancer and has far-reaching therapeutic implications as >70% of breast tumors express ER.Citation1 ER is best characterized as a transcription regulator activated by its ligand estrogen binding, which in turn binds directly or indirectly to DNA in the genome and modulates gene expression involved in normal cell function and tumor progression.Citation2,Citation3 Limiting estrogen binding to ER has been a successful strategy to treat ER-positive (ER+) breast cancer, specifically in an adjuvant setting for the prevention of tumor recurrence. Selective ER modulators (SERMs; eg, tamoxifen) that compete with estrogen for binding to ER have proven effective in reducing tumor recurrence and mortality in ER+ breast cancer patients by 50% and 30%, respectively.Citation4 Newer generation antiestrogen agents, aromatase inhibitors (AIs; eg, anastrozole) that inhibit estrogen production in peripheral tissue, have demonstrated increased efficacy in reducing tumor recurrence in postmenopausal ER+ breast cancer patients.Citation5 These study outcomes have warranted antiestrogen agents as the staple therapy in ER+ breast cancer treatment. However, it has long been recognized that 2%–5% of patients relapse each year even during therapy, indicating that a significant portion of breast tumors have intrinsic resistance to antiestrogen therapy.Citation5
The mechanisms of antiestrogen therapy resistance have been extensively investigated using cell models subjected to long-term hormone deprivation or tamoxifen treatment. One of the converging mechanisms from these studies is aberrant growth factor (GF) signaling involving receptor kinases such as epidermal growth factor receptor (EGFR), ERBB2/HER2, insulin-like growth factor receptor (IGF1R), and fibroblast growth factor receptor 1 (FGFR1), and their downstream molecules such as PI3K/AKT, MAPK, and MYC.Citation6–Citation12 GF signaling not only promotes tumor cell proliferation and survival but also directly activates ER through phosphorylation.Citation6 GF-activated ERα was shown to bind to the genomic regions distinct from estrogen-induced ERα binding sites and induces gene expression related to endocrine therapy resistance.Citation13 Supportive of the notion that GF signaling plays a significant role in aggressive breast cancer and/or therapy resistance, breast tumor samples from patients who do not benefit from therapy often harbor mutations or activation signatures related to the GF signaling pathways.Citation14 Not only does GF signaling activate ER, but a subset of estrogen-bound ER localized in the cytoplasm has also been shown to activate GF signaling by directly associating with GF receptors or with GF signaling components, underscoring the bidirectional crosstalk between ER and GF signaling in breast cancer.Citation6 However, combination therapy using antiestrogen and GF pathway-targeting agents has not been successful in improving breast cancer outcome in clinical trials, presenting challenges to the strategies to counter GF signaling-related endocrine therapy resistance in breast cancer.Citation15–Citation17 The fact that GF signaling pathway mutations have been found in tumors resistant to cancer therapeutics not related to antiestrogen agents may suggest that GF signaling may provide therapy resistance in general by providing tumor cell growth and survival advantage, rather than conferring resistance specific to antiestrogen therapy.Citation18
The more direct mechanism of antiestrogen therapy resistance is mutation of the ER encoding gene, ESR1, found in 20%–50% of recurrent tumors in patients treated with adjuvant SERM and/or AI therapy.Citation19,Citation20 These mutations result in an ER protein conformation that mimics the estrogen-bound state of ER, negating the requirement for estrogen and rendering antiestrogen agents ineffective.Citation21 However, the ESR1 mutations are found at very low frequencies in primary tumors, suggesting that these mutations are likely to represent acquired resistance under selective pressure of antiestrogen therapy.Citation21,Citation22 Thus, the ESR1 mutation status has limited utility as a diagnostic marker and/or therapy target for the antiestrogen therapy resistance that plagues patients during the initial stages of breast cancer treatment.
Several gene expression signatures associated with poor prognosis related to tamoxifen or AI resistance have emerged, some of which are clinically available as prognostic tests.Citation14 However, the variability between the gene signatures may attest to the heterogeneity of intrinsic antiestrogen resistance and/or the diversity of technical and computational platforms used in deriving each gene signature. Clinical utility of the gene signatures to predict resistance to antiestrogen therapy awaits reports from ongoing trials.Citation14 As such, a need to better understand genetic factors that determine intrinsic antiestrogen therapy resistance still remains.
Inhibitor of growth 4 (ING4) is a member of the ING tumor suppressor family (ING1–5) that regulates histone modification and gene transcription.Citation23 It has been shown that the ING4 gene is deleted in 16% or downregulated in 34% of breast tumors.Citation24,Citation25 Low expression of ING4 was correlated with advanced tumor features and lymph node positivity, suggesting that downregulation of ING4 may contribute to breast cancer progression.Citation25 More clinically relevant, patients with ING4-low expressing primary tumors relapsed at a faster rate. In particular, ING4-low expression was associated with more than three times the recurrence rate in a cohort of ER+ breast cancer patients who were treated with adjuvant tamoxifen.Citation25 These results raised a question whether ING4 played a role in ER signaling and/or tamoxifen response. This study investigated a functional relationship between ING4 and ER in breast cancer cells. The results demonstrate that ING4 inhibits ligand-independent ER activity in the nucleus that allows growth of ER+ breast cancer cells in the absence of estrogen. These results suggest that ING4-low tumors contain unregulated ligand-independent ER activity, which renders tamoxifen less effective in patients. This study proposes downregulation of ING4 as a mechanism of intrinsic antiestrogen therapy resistance in ER+ breast cancer.
Materials and methods
Cell culture and reagents
T47D and MCF7 cells that express the retroviral vector pMIG or the pMIG-based ING4 overexpression construct have been previously described.Citation25,Citation26 T47D and MCF7 cells were grown in the Roswell Park Memorial Institute (RPMI) and Minimum Essential Medium with Earle’s Balanced Salt Solution (MEM/EBSS) media (Hyclone, Logan, UT, USA), respectively, containing 10% fetal bovine serum (FBS, Hyclone) and 10 μg/mL human insulin (Sigma-Aldrich, St. Louis, MO, USA). For hormone deprivation, cells were grown in respective phenol red free media (Invitrogen, Carlsbad, CA, USA) containing 10% charcoal-stripped FBS (Hyclone). The reagents 17β-estradiol (E2, Sigma) and ICI182,780 (Sigma) were dissolved in dimethyl sulfoxide (DMSO), and 4-hydroxy tamoxifen (OHT, Sigma) was dissolved in 100% ethanol.
In vitro cell proliferation assay
Cells were plated at a density of 2,000 cells per well in 96-well plates in triplicate wells. Cells were grown in various media conditions for 7–14 days. Cells were fixed with 10% trichloroacetic acid followed by sulforhodamine B (SRB) colorimetric assay to measure relative cell numbers as described previously.Citation25 Cell growth assays were repeated in three or more independent experiments.
Western blot analysis
Cell lysates were fractionated by lysing cells in a hypotonic buffer (10 mM Tris pH 8, 10 mM NaCl, 0.2% Nonidet P-40) on ice for 5 min, followed by centrifugation at 1,800 ×g for 5 min to collect nuclei and cytoplasm. Nuclei were lysed in radioimmunoprecipitation assay (RIPA) buffer followed by sonication. Nuclear and cytoplasmic fractions were analyzed by Western blot using antibodies against ERα (Cell Signaling, Danvers, MA, USA), ING4 (EMD Millipore, Temecula, CA, USA), histone H3 (Cell Signaling), and tubulin (Cell Signaling), and phospho-extracellular signal-regulated kinase (ERK) (Cell Signaling) at 1:1,000 dilution.
Luciferase assay
The luciferase reporter plasmid, 3xERE-TATA-luc, was purchased from Addgene (Cambridge, MA, USA).Citation27 T47D-pMIG or T47D-ING4 cells were co-transfected with the linearized luciferase reporter plasmid and a neomycin resistance gene containing plasmid, pLNCx (Clontech, Mountain View, CA, USA) using Effectene (Qiagen Valencia, CA, USA) and were selected in the media containing 400 mg/mL Geneticin (Gibco, Billings, MT, USA). Cells plated at 50% confluency in a 24-well dish were hormone-deprived in the media containing 10% of charcoal-stripped FBS for 48 h and with or without 100 nM E2 for additional 24 h. Luciferase activity in 30 μL of cell lysates was measured using a Steady-Glo Luciferase Assay System (Promega Corporation, Fitchburg, WI, USA) and Victor3 luminometer (Perkin Elmer Life Sciences Products, Boston, MA, USA). Total protein concentration in cell lysates was measured using the BCA Protein Assay Kit (Pierce Thermo Fisher Scientific, Waltham, MA, USA). Luciferase activity was calculated as relative light units per microgram of protein and normalized to the luc2 gene copy number integrated into the genome as described previously.Citation25
Quantitative reverse transcription polymerase chain reaction (qRTPCR)
Total RNA was isolated from 5×105 cells using the RNeasy mini kit (Qiagen). Complementary DNA was synthesized from 2–5 μg of total RNA using the Superscript III First-Strand Synthesis SuperMix kit with oligo dT primers (Invitrogen). Quantitative PCR was performed using Taqman Gene Expression Assays (Applied Biosystems, Foster City, CA, USA) with FAM-labeled probes for PDZK1, TFF1, ESR1, PGR, and MYC, and VIC-labeled probe for GAPDH. Reactions were run using the Applied Biosystems 7900HT (Applied Biosystems) or StudioQuant 6 Real-Time PCR instrument (Life Technology, Carlsbad, CA, USA). Data were analyzed by the ΔΔCt method normalized to GAPDH.
Statistical analysis
Unpaired 2-tailed Student’s t-test was used to determine the statistical significance between two experimental number sets. Estrogen dose–response curve and half-maximal effective concentration (EC50) values were generated by nonlinear regression analysis with variable slope using GraphPad Prism software version v6.0 (GraphPad Software, Inc., La Jolla, CA, USA). Extra sum of squares F-test was used to determine a statistical significance of the differences between the two curves.
Results
ING4 overexpression increases sensitivity of ER+ breast cancer cells to hormone deprivation and to tamoxifen treatment
To assess the effects of ING4 on estrogen signaling, ING4 was overexpressed in two ER+ breast cancer cell lines, T47D and MCF7, using the retroviral vector pMIG as described previously.Citation25 The growth between the vector (pMIG) and ING4 overexpressing cells was compared in the media containing full serum (FS) or in the hormone-deprived media containing charcoal-stripped serum (CSS) with or without E2. The ability of OHT blocking E2-depedent cell growth was also assessed in the same assay. The results showed that ING4 cells grew with a comparable rate to pMIG cells in FS, as has been reported previously;Citation25,Citation26 pMIG or ING4 cells increased cell numbers by 200%–300% in 7 days (). In CSS, pMIG cells grew by ≤50% in 7 days (), demonstrating that both T47D and MCF7 cells were sensitive to hormone deprivation but capable of growing in the absence of hormones albeit with a 4- to 6-fold reduced rate. In comparison, ING4 overexpressing T47D cells did not increase in cell numbers in CSS (). Moreover, MCF7 cells overexpressing ING4 showed not only no growth but also cell death by 50% (). These results indicated that ING4 overexpression resulted in increased sensitivity to hormone deprivation and/or suppressed hormone-independent growth of T47D and MCF7 ER+ breast cancer cells.
Figure 1 ER+ breast cancer cells overexpressing ING4 are more sensitive to hormone deprivation and to tamoxifen treatment.
Abbreviations: ER+, estrogen receptor-positive; ING4, inhibitor of growth 4; CSS, charcoal-stripped serum; FS, full serum; E2, 17β-estradiol; OHT, 4-hydroxy tamoxifen; SRB, sulforhodamine B.
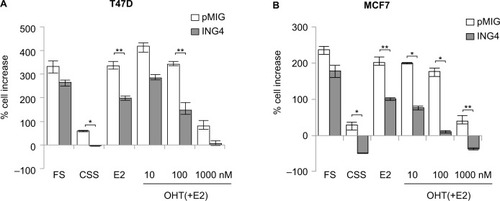
In the presence of E2 in CSS, pMIG cells grew with a rate comparable to the cells in FS, indicating that estrogen alone could reconstitute growth signaling in the absence of other hormones (). In contrast, ING4 cells had reduced rates of cell growth in the presence of E2 compared to pMIG cells, indicating that ING4 overexpression attenuated estrogen signaling for the growth of T47D and MCF7 cells (p<0.002 by Student’s t-test; ). Consistent with these findings, ING4 overexpressing cells were more sensitive to OHT compared to pMIG cells (). At 1 μM concentration of OHT, the cell growths were identical to the ones in CSS for pMIG or ING4 cells, indicating that 10 nM of E2 was effectively blocked by 1 μM OHT (). These results indicated that ING4 increased the sensitivity of T47D and MCF7 cells to tamoxifen by attenuating estrogen-dependent growth and/or by suppressing hormone-independent growth.
ING4 attenuates estrogen signaling for cell growth without affecting estrogen dose–response
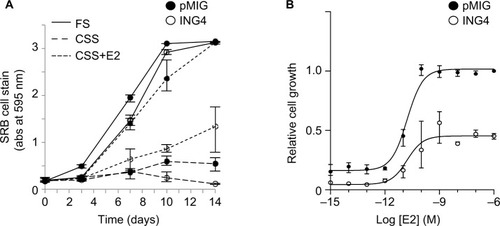
In order to determine whether growth retardation of ING4 cells in E2 was due to altered estrogen response, T47D cell growth rates were compared between pMIG and ING4 cells over a 14-day time period in the presence or absence of E2. The results showed that ING4 cells grew at a similar rate as pMIG cells in FS, whereas ING4 cells grew at a 2–3 times reduced rate in E2 compared to pMIG cells (2.7× slower rate shown in ). These results suggested that ING4 may have suppressed E2 signaling for cell growth, potentially requiring higher dose of E2 to achieve optimal growth signaling. EC50 analysis was carried out to determine whether ING4 cells required a higher dosage of estrogen for growth comparable to pMIG cells. The results showed that the EC50 for estrogen in pMIG cells was 15 pM while it was 32.9 pM for ING4 cells (). The difference between the two EC50 values was not statistically significant, indicating that the estrogen dose–response was comparable between pMIG and ING4 cells. However, the difference between the two dose–response curves was statistically significant (p<0.0001 by extra sum of squares F-test; ), indicating that ING4 attenuated the overall E2-mediated cell growth without affecting estrogen dose–response. These results suggested that ING4 functions as a noncompetitive inhibitor of estrogen signaling, potentially by reducing ER protein levels and/or by inhibiting estrogen-independent ER activity.
Figure 2 ING4 retards estrogen-dependent growth of T47D cells.
Abbreviations: ING4, inhibitor of growth 4; CSS, charcoal-stripped serum; FS, full serum; E2, 17β-estradiol; SRB, sulforhodamine B; EC50, half-maximal effective concentration.
ING4 does not affect nuclear ERα protein expression
To determine whether ING4 overexpression resulted in reduction of the ERα protein, Western blot analysis was used. T47D or MCF7 cells expressing pMIG or ING4 were incubated in FS, CSS, CSS+E2, or CSS+E2+OHT, for 24 h. Cells were also treated with fulvestrant ICI182,780 (ICI) in CSS+E2 media for 24 h. Cell lysates were fractionated to nuclear and cytosolic portions and blotted for ERα. The results showed that the amounts of the ERα protein in the nuclear fraction were virtually identical between pMIG and ING4 cells (). ERα protein levels in the nuclear fraction decreased when cells were deprived of hormones (CSS), while they increased in the presence of E2, reflecting that ING4 did not influence nuclear localization of ERα upon E2 binding. OHT increased nuclear accumulation of ERα, whereas ICI treatment reduced amounts of the ERα protein via enhanced degradation as described previously.Citation28 These results demonstrated that ING4 did not reduce nuclear ERα protein levels or nuclear localization of ligand-bound ERα, suggesting that ING4 may inhibit the ER activity instead.
Figure 3 ING4 inhibits ERα transcription activity without affecting ERα protein expression.
Abbreviations: ING4, inhibitor of growth 4; CSS, charcoal stripped serum; FS, full serum; E2, 17β-estradiol; OHT, 4-hydroxy tamoxifen; ICI, ICI182,780; ER, estrogen receptor; qRTPCR, quantitative reverse transcription polymerase chain reaction; ERK, extracellular signal-regulated kinase.
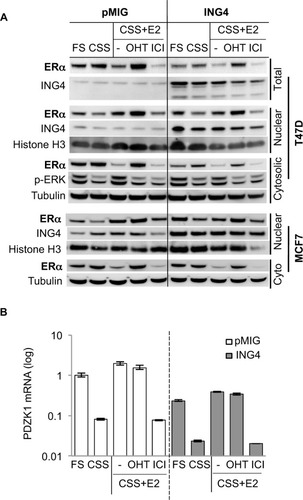
It is notable that the ERα protein levels in the cytosolic fractions were reduced by ~2-fold in T47D cells overexpressing ING4, which was also reflected in the total lysate (). However, E2-induced phosphorylated ERK was comparable between pMIG and ING4 cells, indicating that the non-genomic function of ER involving the MAPK signaling pathway in the cytosol was not compromised (). In addition, MCF7 cells overexpressing ING4 showed comparable amounts of the ERα protein in the cytosolic and nuclear fractions to the pMIG control cells (, bottom panel). Thus, it is unlikely that ING4 regulates ERα protein expression, and that the reduction in cytoplasmic ERα contributes to the increased sensitivity of T47D-ING4 cells to hormone deprivation.
ING4 represses an ER-target gene, PDZK1
As the Westen blot results showed that the amounts of nuclear ERα protein were comparable between pMIG and ING4 cells, ER-mediated transcription was investigated next, by measuring. mRNA expression of one of the ER-target genes, PDZK1, in T47D cells incubated in the same media conditions of the Western blot samples in , using quantitative RTPCR assays. The results showed that in pMIG cells, PDZK1 expression increased by 12-fold and 24-fold in FS and E2, respectively, compared to the cells in CSS (; open bars). Unexpectedly, E2-induced PDZK1 expression was not inhibited by OHT, but was completely inhibited by ICI (). These results suggested that PDZK1 expression may largely be mediated by E2-independent ER.
In ING4 cells, PDZK1 expression was increased by 10-fold and 17-fold in FS and E2, respectively, compared to the cells in CSS (; solid bars), indicating that ING4 did not affect E2-induced expression of PDZK1. As was in pMIG cells, E2-induced expression of PDZK1 was inhibited by ICI but not by OHT in ING4 cells, consistent with E2-independent ER-mediated expression of PDZK1. It was apparent however, that PDZK1 expression levels were reduced in ING4 cells in all media conditions by 4- to 5-fold, compared to the ones in pMIG cells (). These results indicated that ING4 repressed “basal level” expression of PDZK1, potentially by inhibiting genomic action of E2-independent ER.
Fulvestrant, not tamoxifen, inhibits the basal PDZK1 expression
Next was tested whether PDZK1 expression in pMIG cells could be reduced to the level in ING4 cells by inhibiting ER. pMIG cells were treated in FS, CSS, or CSS+E2, and PDZK1 expression was compared in the presence or absence of tamoxifen (OHT) or fulvestrant (ICI), compared to the PDZK1 expression in ING4 cells. The results showed that OHT did not, but ICI did reduce PDZK1 expression in FS and E2, suggesting that PDZK1 expression was E2-independent but ER-dependent (). In FS, ING4 reduced the expression level of PDZK1 comparable to ICI-treated pMIG cells, indicating that ING4 too inhibited E2-independent ER activity (). In E2, ING4 reduced PDZK1 expression but to a lesser degree compared to ICI-treated pMIG cells, suggesting that E2 may override ING4-mediated PDZK1 repression (). In CSS, ICI treatment did not further reduce PDZK1 expression, suggesting that the “basal level” expression of PDZK1 may be ER-independent. In contrast, ING4 reduced PDZK1 expression in CSS, suggesting that ING4 may additionally inhibit ER-independent expression of PDZK1 (see “Discussion”). It is notable that OHT increased PDZK1 expression in CSS, indicating an agonist effect of OHT in the absence of E2 (). Collectively, these results demonstrated that while OHT did not, both ICI and ING4 decreased PDZK1 expression, indicating that ING4 inhibited E2-independent ER.
Figure 4 ING4 inhibits ligand-independent ER activity.
Abbreviations: ING4, inhibitor of growth 4; CSS, charcoal stripped serum; FS, full serum; E2, 17β-estradiol; OHT, 4-hydroxy tamoxifen; ER, estrogen receptor; ICI, ICI182,780; qRTPCR, quantitative reverse transcription polymerase chain reaction.
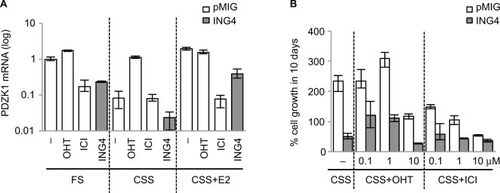
Fulvestrant, but not tamoxifen, inhibits hormone-independent cell growth of T47D cells
As ING4 inhibited E2-independent ER activity that mediated PDZK1 expression, it was investigated whether ING4 inhibition of E2-independent cell growth was ER-dependent. pMIG cell growth in CSS in the presence of OHT or ICI was compared to ING4 cell growth. The results showed that pMIG cell number increased 200% in 10 days in CSS, demonstrating hormone-independent growth (). In contrast, ING4 cell number was decreased to 50%, indicating cell death in prolonged hormone deprivation conditions (). Next, increasing log doses of OHT or ICI were used in CSS and the percent cell growth of pMIG cells was compared to ING4 cell growth in CSS. The results showed that OHT at 0.1 or 1 μM concentration did not inhibit pMIG cell growth in CSS. The same concentrations of OHT stimulated the growth of ING4 cells in CSS, indicating an agonist effect of OHT in cell growth in the absence of E2. These findings are consistent with the results showing that OHT induced PDZK1 expression in the absence of E2 (). In the presence of 10 μM OHT, both pMIG and ING4 cells showed cell death, reflecting a cytotoxic effect of OHT at a high concentration (). In contrast, ICI inhibited growth of pMIG cells in a dose-dependent manner, with 10 μM concentration effectively suppressing hormone-independent growth of pMIG cells to the level of ING4 cells (). These results demonstrated that ICI but not OHT rendered pMIG cells sensitive to hormone-deprivation as ING4 cells. These results support the conclusion that ING4 inhibits E2-independent ER activity that allows ER+ breast cancer cells to grow in the absence of estrogen.
ING4 inhibits the expression of estrogen responsive element (ERE)-mediated reporter luciferase gene
Next was investigated whether ING4 regulation of ER-target genes involved the ERE sequence, a canonical ER binding site. The pGL2-ERE plasmid containing a luciferase reporter gene with ERE in the promoter region was used to establish T47D-pMIG or T47D-ING4 cell lines stably transfected the reporter construct. Luciferase assay was carried out using cell lysates harvested after incubating cells in CSS for 2 days and additional 24 h with or without E2. The results showed that ING4 cells contained at least 100-fold less luciferase activity in CSS () compared to pMIG cells, indicating that ING4 repressed basal expression of the luciferase reporter gene. E2 treatment induced luciferase activity by 5-fold in pMIG cells but failed to induce it in ING4 cells, indicating that ING4 inhibited E2-induced expression of the reporter gene (). Thus, it was concluded that ING4 inhibited both basal and E2-induced expression of the luciferase reporter gene facilitated by the ERE sequence.
Figure 5 ING4 represses transcription of selective ER-target genes containing ERE.
Abbreviations: ING4, inhibitor of growth 4; CSS, charcoal stripped serum; FS, full serum; E2, 17β-estradiol; ER, estrogen receptor; ERE, estrogen response element.
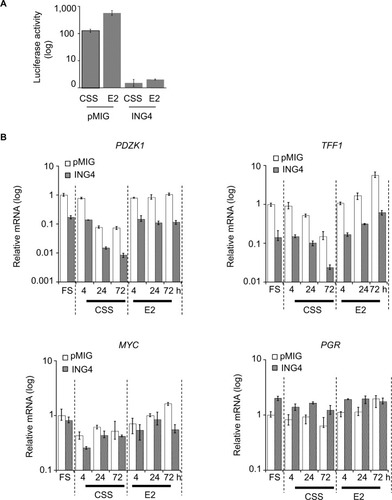
It was notable that transient transfection of the reporter construct did not yield any discernable or reproducible differences in the basal or E2-induced luciferase expression between pMIG and ING4 cells (data not shown). These results indicated that the action of ING4 on transcription modulation required the reporter gene integration in the genome as has been reported previously, consistent with the proposed mechanism of ING4 as a chromatin remodeler.Citation25
ING4 represses selective ER-target gene expression
ING4 regulation of four endogenous ER-target gene expression was next investigated: PDZK1, TFF1, MYC, and PGR. The first two genes, PDZK1 and TFF1, have been shown to contain a canonical ERE in their promoter regions while the latter two, MYC and PGR, have a putative ERE half site.Citation29–Citation32 The results showed that PDZK1 or TFF1 mRNA levels were 10-fold less in ING4 cells in all media conditions compared to pMIG cells (). In contrast, ING4 did not affect MYC or PGR expression (). E2-induced fold expression of all four genes was not or nominally affected by ING4 (). E2-induced expression of MYC seemed to be reduced in ING4 cells at a 72-h time point (). However, a time course experiment showed comparable MYC induction in ING4 cells at a later time point (data not shown). It is unclear whether ING4 plays a role in temporal regulation of MYC expression. Nevertheless, these results indicated that ING4 repressed basal expression of PDZK1 and TFF1, but not of MYC and PGR (). Although limited in scope, since both PDZK1 and TFF1 in addition to the luciferase reporter gene contain the canonical ERE sequence in their promoter regions, these findings may suggest that ING4 regulates basal expression of ER-target genes that contain ERE.
Discussion
The present study has demonstrated that ING4 inhibits estrogen-independent ER activity that sustains growth of ER+ breast cancer cells under hormone-deprived conditions. These results suggest that breast tumors that have downregulated ING4 conversely contain high estrogen-independent ER activity. Consequently, antiestrogen agents such as tamoxifen and AIs would not be effective in the treatment of ING4-low ER+ breast cancer. These findings provide a possible explanation for the previous report that breast cancer patients with ING4-low tumors recur with faster rates despite adjuvant tamoxifen therapy.Citation25 The findings further suggest that blocking estrogen with SERM or AI is not sufficient for the treatment of ING4-low ER+ breast cancer, but selective ER degraders such as fulvestrant that facilitates ER degradation may be more effective in preventing tumor recurrence. Supportive of this idea are the recent clinical trials of fulvestrant showing improved patient survival over AI.Citation33 Whether breast tumors with low ING4 levels show increased response to fulvestrant has not been determined.
The study results put forth ING4 downregulation resulting in disregulated estrogen-independent ER activity as a mechanism for antiestrogen therapy resistance. High levels of ligand-independent ER activity have been detected in AI-resistant breast cancer cells selected in vitro or in patient tumor samples collected after the treatment.Citation34,Citation35 These studies have suggested that ligand-independent ER activity may be an acquired trait under therapy selection. Whether increased ligand-independent ER activity in AI recurrent tumors is in part due to ING4 downregulation is not known, nor is ING4 downregulation an acquired trait under therapy selection. However, it was previously shown that ING4 expression in primary tumors could stratify recurrent patients with tamoxifen therapy,Citation25 indicating that ING4 downregulation had occurred prior to tamoxifen therapy at least in this patient cohort. These results suggest that low ING4 expression may confer intrinsic tamoxifen resistance prior to therapy and further suggest that ING4 expression in primary tumors could be used as a prognostic biomarker predictive of antiestrogen therapy response.
The molecular mechanism of ING4 downregulation in tumors is not well understood. The previous study reported that 16% of primary breast tumors harbor a single copy ING4 gene deletion, which is likely to account for low ING4 expression in a subset of tumors.Citation24 Other mechanisms including epigenetic regulation of ING4 gene expression are currently unknown. Such mechanisms may be potentially targeted by pharmacological means to achieve ING4 induction in order to thereby reduce ligand-independent ER activity and prevent tumor recurrence in breast cancer.
In this study, ING4 was shown to repress the expression of selective ER-target genes, PDZK1 and TFF1, but not MYC and PGR. Whether ING4-repressed ER-target genes including PDZK1 and TFF1 play a functional role in the pathogenesis and/or therapy resistance of ER+ breast cancer is presently unclear. It is notable, however, that PDZK1 has been identified as a breast cancer susceptibility gene in genome-wide association studies.Citation36 Moreover, ectopic expression of PDZK1 has been shown to increase the proliferation of MCF7 ER+ breast cancer cells in the presence or absence of estrogen, suggesting an oncogenic potential of PDZK1.Citation37 Thus, repression of PDZK1 basal expression could be part of ING4’s tumor suppressive mechanism. However, PDZK1 mRNA expression levels alone did not stratify recurrent breast cancer patients with tamoxifen therapy (data not shown), indicating that PDZK1 regulation is not the sole tumor suppressor function of ING4 in breast cancer and/or therapy response.
The scope of ER-target genes that ING4 regulates is currently unclear, as several gene expression-profiling studies have not yielded a consistent model of ING4 action (S Kim, unpublished data). This is likely due to the dynamic nature of ERα transcriptional activity reported in extensive and numerous studies, identifying 300 to >1,000 estrogen-induced ER-target genes.Citation3,Citation38,Citation39 Moreover, studies have identified diverse co-activators and repressors that interact with liganded and unliganded ERα.Citation40–Citation43 The exact molecular mechanism of ING4 in ERα regulation is presently unclear. ING4 and ERα did not co-immunoprecipitate, suggesting indirect and/or transient interactions if any (S Kim, unpublished data). Limited chromatin immunoprecipitation (ChIP) experiments showed that ERα was present on the ERE promoter sequence of the reporter construct in ING4 overexpressing cells, indicating that ING4 did not inhibit ERα binding to the genome (S Kim, unpublished data). However, whether ING4 binds to the same ERE promoter region is undetermined because no evidence is available to confirm that the ING4 antibody is capable of ChIP. Such an antibody will be a crucial reagent to decipher the ING4 mechanism.
Recent studies have also shown that unliganded ERα binds to 4,000–5,000 sites in the genome.Citation35,Citation40,Citation44 Caizzie et al in their study demonstrated that unliganded ERα regulates ~900 genes that largely overlap with the set of estrogen-induced ER-target genes, suggesting that unliganded ERα may maintain the basal expression of estrogen-inducible ER-target genes.Citation40 These results support the current study findings that basal expression of PDZK1 was effectively repressed by fulvestrant but not by tamoxifen. The current study also suggests that ING4 may play a role in the regulation of genes that contain canonical ERE in their promoter regions. To the authors’ knowledge, this study puts forth ING4 as the first negative regulator of unliganded ERα. Caizzi et al also reported that unliganded ERα regulated expression of a distinct set of genes involved in the luminal epithelial cell fate.Citation40 These data would suggest that ING4 may also play a role in breast epithelial differentiation via regulation of unliganded ERα. Supportive of this, expression of a dominant negative mutant of ING4 resulted in hyperplastic abnormal ductal structure in mouse mammary glands and cooperated with the MYC oncogene to form mammary tumors.Citation45 In addition, ING4 was shown to regulate gene transcription during prostate epithelial cell differentiation and that in the absence of ING4, primary prostate epithelial cells progress to cancer with additional oncogenic events.Citation46 Taken together, it can be postulated that ING4 plays a broader transcriptional regulatory role in epithelial differentiation and/or maintenance, thus suppressing tumorigenesis. A more direct approach to determine ING4-target genes and/or ING4 interaction with unliganded ERα in the genome in mammary tissue development will likely clarify the roles of ING4/ERα interplay in mammary epithelial cell differentiation and breast cancer.
Finally, it is notable that low expression of ING4 has been correlated with advanced tumors, poor patient outcome, and therapy resistance in diverse cancers including glioma, head and neck squamous carcinoma, breast cancer, lung cancer, hepatocellular carcinoma, prostate cancer, and multiple myeloma.Citation23 These studies attest to a tumor suppressive function of ING4 in many tissue types, not all of which express ERα. Therefore, inhibition of unliganded ERα may represent one of multiple functions of ING4. Indeed, ING4 has been shown to regulate other transcriptional programs related to p53 in DNA damage response, NFκB in tumor progression and immune response, and Myc in prostate cell differentiation and mammary tumorigenesis.Citation23 The molecular mechanism of ING4 that enables its interactions with diverse transcriptional factors including unliganded ERα will need to be determined in order to address the fundamental function and mechanism of ING4 in normal and cancer cell development and also to explore a potential innovative strategy to treat cancer.
Disclosure
The authors report no conflicts of interest in this work.
Acknowledgments
The authors thank Dr Sara Byron for her critical reading of the manuscript and helpful discussions. The current address of Madeline M Keenen is University of California, San Francisco, CA, USA (Email [email protected]). This work was supported by funding from the University of Arizona College of Medicine-Phoenix (to Suwon Kim).
References
- AndersonWFChatterjeeNErshlerWBBrawleyOWEstrogen receptor breast cancer phenotypes in the Surveillance, Epidemiology, and End Results databaseBreast Cancer Res Treat2002761273612408373
- ShaoWBrownMAdvances in estrogen receptor biology: prospects for improvements in targeted breast cancer therapyBreast Cancer Res200461395214680484
- WelborenWJSweepFCSpanPNStunnenbergHGGenomic actions of estrogen receptor alpha: what are the targets and how are they regulated?Endocr Relat Cancer20091641073108919628648
- DaviesCGodwinJGrayREarly Breast Cancer Trialists’ Collaborative GroupRelevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: patient-level meta-analysis of randomised trialsLancet2011378979377178421802721
- DowsettMForbesJFBradleyREarly Breast Cancer Trialists’ Collaborative GroupAromatase inhibitors versus tamoxifen in early breast cancer: patient-level meta-analysis of the randomised trialsLancet2015386100011341135226211827
- OsborneCKSchiffRMechanisms of endocrine resistance in breast cancerAnnu Rev Med20116223324720887199
- ShouJMassarwehSOsborneCKMechanisms of tamoxifen resistance: increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancerJ Natl Cancer Inst2004961292693515199112
- FoxEMMillerTWBalkoJMA kinome-wide screen identifies the insulin/IGF-I receptor pathway as a mechanism of escape from hormone dependence in breast cancerCancer Res201171216773678421908557
- TurnerNPearsonASharpeRFGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancerCancer Res20107052085209420179196
- MartinLAFarmerIJohnstonSRAliSMarshallCDowsettMEnhanced estrogen receptor (ER) alpha, ERBB2, and MAPK signal transduction pathways operate during the adaptation of MCF-7 cells to long term estrogen deprivationJ Biol Chem200327833304583046812775708
- MillerTWHennessyBTGonzalez-AnguloAMHyperactivation of phosphatidylinositol-3 kinase promotes escape from hormone dependence in estrogen receptor-positive human breast cancerJ Clin Invest201012072406241320530877
- MillerTWBalkoJMGhazouiZA gene expression signature from human breast cancer cells with acquired hormone independence identifies MYC as a mediator of antiestrogen resistanceClin Cancer Res20111772024203421346144
- LupienMMeyerCABaileySTGrowth factor stimulation induces a distinct ER(alpha) cistrome underlying breast cancer endocrine resistanceGenes Dev201024192219222720889718
- MaCXReinertTChmielewskaIEllisMJMechanisms of aromatase inhibitor resistanceNat Rev Cancer201515526127525907219
- JohnstonSPippenJJrPivotXLapatinib combined with letrozole versus letrozole and placebo as first-line therapy for postmenopausal hormone receptor-positive metastatic breast cancerJ Clin Oncol200927335538554619786658
- OsborneCKNevenPDirixLYGefitinib or placebo in combination with tamoxifen in patients with hormone receptor-positive metastatic breast cancer: a randomized phase II studyClin Cancer Res20111751147115921220480
- RobertsonJFFerreroJMBourgeoisHGanitumab with either exemestane or fulvestrant for postmenopausal women with advanced, hormone-receptor-positive breast cancer: a randomised, controlled, double-blind, phase 2 trialLancet Oncol201314322823523414585
- KimSNew and emerging factors in tumorigenesis: an overviewCancer Manag Res2015722523926251629
- RobinsonDRWuYMVatsPActivating ESR1 mutations in hormone-resistant metastatic breast cancerNat Genet201345121446145124185510
- ToyWShenYWonHESR1 ligand-binding domain mutations in hormone-resistant breast cancerNat Genet201345121439144524185512
- ThomasCGustafssonJAEstrogen receptor mutations and functional consequences for breast cancerTrends Endocrinol Metab201526946747626183887
- WangPBahreiniAGyanchandaniRSensitive detection of mono- and polyclonal ESR1 mutations in primary tumors, metastatic lesions, and cell-free DNA of breast cancer patientsClin Cancer Res20162251130113726500237
- CuiSGaoYZhangKChenJWangRChenLThe emerging role of inhibitor of growth 4 as a tumor suppressor in multiple human cancersCell Physiol Biochem201536240942225968091
- TapiaCZlobecISchneiderSDeletion of the inhibitor of growth 4 (ING4) tumor suppressor gene is prevalent in human epidermal growth factor 2 (HER2)-positive breast cancerHum Pathol201142798399021315418
- ByronSAMinEThalTSNegative regulation of NF-kappaB by the ING4 tumor suppressor in breast cancerPLoS One2012710e4682323056468
- KimSChinKGrayJWBishopJMA screen for genes that suppress loss of contact inhibition: identification of ING4 as a candidate tumor suppressor gene in human cancerProc Natl Acad Sci U S A200410146162511625615528276
- HallJMMcDonnellDPThe estrogen receptor beta-isoform (ERbeta) of the human estrogen receptor modulates ERalpha transcriptional activity and is a key regulator of the cellular response to estrogens and antiestrogensEndocrinology1999140125566557810579320
- LupienMJeyakumarMHebertERaloxifene and ICI182,780 increase estrogen receptor-alpha association with a nuclear compartment via overlapping sets of hydrophobic amino acids in activation function 2 helix 12Mol Endocrinol200721479781617299137
- TachibanaKAnzaiNUedaCRegulation of the human PDZK1 expression by peroxisome proliferator-activated receptor alphaFEBS Lett2008582283884388818955051
- SewackGFHansenUNucleosome positioning and transcription-associated chromatin alterations on the human estrogen-responsive pS2 promoterJ Biol Chem19972724931118311299388265
- DubikDShiuRPMechanism of estrogen activation of c-myc oncogene expressionOncogene199278158715941630819
- PetzLNZieglerYSSchultzJRKimHKemperJKNardulliAMDifferential regulation of the human progesterone receptor gene through an estrogen response element half site and Sp1 sitesJ Steroid Biochem Mol Biol200488211312215084343
- RobertsonJFLlombart-CussacARolskiJActivity of fulvestrant 500 mg versus anastrozole 1 mg as first-line treatment for advanced breast cancer: results from the FIRST studyJ Clin Oncol200927274530453519704066
- MillerTWBalkoJMFoxEMERalpha-dependent E2F transcription can mediate resistance to estrogen deprivation in human breast cancerCancer Discov20111433835122049316
- VareslijaDMcBryanJFaganAAdaptation to AI therapy in breast cancer can induce dynamic alterations in ER activity resulting in estrogen-independent metastatic tumorsClin Cancer Res201622112765277726763249
- MichailidouKBeesleyJLindstromSGenome-wide association analysis of more than 120,000 individuals identifies 15 new susceptibility loci for breast cancerNat Genet201547437338025751625
- KimHAbd ElmageedZYJuJPDZK1 is a novel factor in breast cancer that is indirectly regulated by estrogen through IGF-1R and promotes estrogen-mediated growthMol Med20131925326223821363
- CarrollJSMeyerCASongJGenome-wide analysis of estrogen receptor binding sitesNat Genet200638111289129717013392
- LinCYVegaVBThomsenJSWhole-genome cartography of estrogen receptor alpha binding sitesPLoS Genet200736e8717542648
- CaizziLFerreroGCutrupiSGenome-wide activity of unliganded estrogen receptor-alpha in breast cancer cellsProc Natl Acad Sci U S A2014111134892489724639548
- ShangYHuXDiRenzoJLazarMABrownMCofactor dynamics and sufficiency in estrogen receptor-regulated transcriptionCell2000103684385211136970
- MetivierRPenotGHubnerMREstrogen receptor-alpha directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoterCell2003115675176314675539
- CarrollJSLiuXSBrodskyASChromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1Cell20051221334316009131
- StellatoCPorrecaICuomoDTaralloRNassaGAmbrosinoCThe “busy life” of unliganded estrogen receptorsProteomics201616228830026508451
- KimSWelmALBishopJMA dominant mutant allele of the ING4 tumor suppressor found in human cancer cells exacerbates MYC-initiated mouse mammary tumorigenesisCancer Res201070125155516220501848
- BergerPLFrankSBSchulzVVTransient induction of ING4 by Myc drives prostate epithelial cell differentiation and its disruption drives prostate tumorigenesisCancer Res201474123357336824762396