Abstract
Although metastatic breast cancer remains essentially incurable, many patients previously treated with an anthracycline, taxane, and capecitabine are relatively fit and keen to receive further therapy. Several drugs are used in this setting, but with little evidence of clinically relevant benefit, and none have previously shown improved survival. Eribulin (Halaven®) is a nontaxane tubulin-binding agent with a novel mode of action, and was recently approved by the European Medicines Agency and US Food and Drug Agency as a single agent for patients with heavily pretreated metastatic breast cancer. This review provides an overview of the discovery, and preclinical and clinical development of eribulin, culminating in the recently published EMBRACE metastatic breast cancer study.
Introduction
Breast cancer is the leading cause of cancer-related death for women in Europe, and second only to lung cancer in the US. Over recent years, breast cancer diagnoses have increased, but mortality has fallen steadily due to a combination of screening, as well as adjuvant endocrine, chemotherapy, and, most recently, HER-2 directed therapy. Only 5% of patients with breast cancer have overt metastatic disease at diagnosis, but at least 30% of those initially diagnosed with early breast cancer will later relapse.Citation1,Citation2 Survival has also improved for these women with metastatic breast cancer,Citation3 with a 5-year survival rate of about 23%,Citation1 but metastatic breast cancer remains essentially incurable.
In both the adjuvant and metastatic settings, molecular markers, historically the estrogen/progesterone receptor and more recently HER-2 status, influence the use of endocrine therapy and HER-2-directed agents, respectively. However, chemotherapy remains at the heart of breast cancer therapy, not only for patients with estrogen/progesterone receptor-negative and HER-2 negative (triple negative) disease, who have an especially poor prognosis,Citation4 but also for those with estrogen/progesterone receptor-positive disease no longer amenable to endocrine therapy or as a partner for HER-2 targeted therapy in patients with HER-2 positive disease.
The best established chemotherapeutic agents for breast cancer are the anthracyclines, taxanes, and capecitabine, an oral fluoropyrimidine. Initially used principally in the metastatic setting, the former two are increasingly part of adjuvant regimens.Citation5–Citation8 Women with metastatic breast cancer who have received an anthracycline, taxane, and capecitabine often remain relatively fit and keen for further therapy, but their prior therapy limits the options.Citation9 Several drugs have shown some activity in Phase II trials, including ixabepilone,Citation10 vinorelbine,Citation11 gemcitabine,Citation12 pemetrexed,Citation13 and the platinums.Citation14 In addition, the value of retreatment with an agent to which the patient has previously been exposed remains unclear. Capecitabine and ixabepilone are approved as monotherapy (the latter in the US and some other countries) in patients with metastatic breast cancer already treated with anthracyclines and taxanes,Citation15 but neither has shown survival benefit in this setting. Therefore, there has been no robust evidence base or “standard of care” for these women.
Eribulin mesylate (Halaven®) is a nontaxane tubulin-binding agent with a novel mode of action, and was recently approved by the European Medicines Agency and US Food and Drug Administration. Encouraging Phase IICitation42–Citation44 data have been supplemented by recent Phase IIICitation45 data showing eribulin to be the first monotherapy to prolong survival in women with heavily pretreated metastatic breast cancer.
Preclinical development
Discovery and synthesis
Eribulin is derived from the marine environment, a potentially rich but challenging source of novel natural products as anticancer drugs, with around 592 marine compounds having shown cytotoxic and antitumor activity.Citation16
In 1986, Hirata and Nemura isolated halichondrin B, a polyether macrolide, from the rare marine sponge, Halichondria okadai ().Citation17 This structurally complex compound had considerable tubulin-mediated antitumor activity in vivo and in vitro.Citation18 During the 1990s, the US National Cancer Institute led attempts at aquaculture, but these were ultimately unsuccessful.Citation19
Figure 1 Halichondria okadai, Halichondrin B and eribulin mesylate.
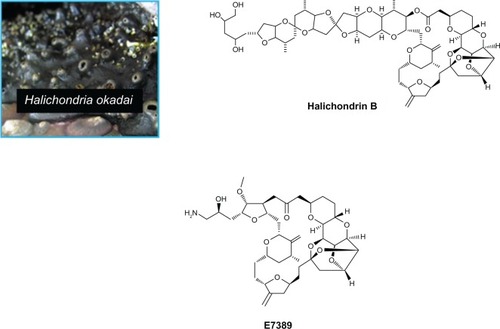
The total synthesis of halichondrin B was achieved by Kishi and colleagues at Harvard in 1992, and Eisai, a Japanese pharmaceutical company, began testing it as an anticancer agent.Citation20 Biological activity was shown to reside in the macrocyclic lactone C1–C8 moiety, located on the right half of the molecule. In a collaboration between academia and industry, a series of analogs that retained the pharmacophore was synthesized and evaluated. In 1997, eribulin mesylate (E7389, ) was synthesized, retaining the cytotoxicity of halichondrin B as well as being water-soluble and chemically stable. Eribulin is structurally much simpler than the parent compound, but is still said to be the most structurally complex fully synthetic molecule to be approved.Citation21–Citation23
Pharmacodynamics and mode of action
The cytotoxicity of eribulin is mediated through its action on what can be considered a “validated” target, tubulin within the mitotic spindle. However, eribulin is distinct from other agents interacting with tubulin (ie, the vinca alkaloids, taxanes, and epothilones) with respect to its chemical structure and the nature of the interaction with tubulin.Citation24,Citation25
Eribulin binds specifically, in a concentration-dependent manner and with high affinity, to a limited number of tubulin sites on the (+) ends of the microtubule. This is distinct from the vinca alkaloids that bind to the (+) ends and also along the sides of the microtubule. Likewise, although there are subtle differences in their mode of binding, the taxanes and epothilone B both bind to β tubulin subunits on the inside of the microtubule.Citation25,Citation26 These differences are reflected in eribulin inhibiting (polymerization) but not shortening (depolymerization) microtubule growth. In contrast, the vinca alkaloids, taxanes, and epothilones all inhibit both microtubule growth and shortening.
Eribulin differs from other agents that interact with tubulin in further ways. Firstly, eribulin promotes centromere spindle relaxation without affecting the rate of stretching. Secondly, eribulin sequesters tubulin into nonfunctional aggregates that can compete with soluble tubulin for addition to the end of growing microtubules.
As a result of its effects on the microtubule, eribulin causes arrest in the G2/M phase of the cell cycle and prolonged mitotic blockade, with marked disruption of normal mitotic spindle architecture, leading to activation of the apoptotic cascade and cell death.Citation18,Citation27–Citation29
Preclinical activity, toxicity, and pharmacokinetics
Eribulin has demonstrated activity both in vitro and in vivo, with a relatively wide therapeutic window and favorable pharmacokinetics. Eribulin was active at nanomolar concentrations against lung, ovary, prostate, colon, and also breast cancer cell lines, including MDA-MB-435, where eribulin was more potent than either vinblastine or paclitaxel.Citation18 Notably, eribulin retained full activity in ovarian cell lines with β-tubulin mutations resistant to paclitaxel.Citation30 In another study, only βIII tubulin expression levels appeared to correlate with sensitivity to eribulin,Citation31 and this relationship was not sufficiently powerful to be used as a biomarker strategy in the clinic.
Human xenograft studies of a range of tumor types in vivo, including three breast cancer models, showed activity at doses of 0.05–1.0 mg/kg. Eribulin was more potent than paclitaxel and the vinca alkaloids,Citation18 again with striking activity against MDA-MB-435 breast cancer xenografts. In preclinical models, intermittent dosing was less toxic and more effective than daily dosing.Citation32 Eribulin also had a wide therapeutic window; specifically, eribulin appeared to cause less neurotoxicity, manifest as less functional and morphological damage, than paclitaxel in mice.Citation33
Preclinical pharmacokinetic studies in rats showed extensive tissue distribution, and a prolonged elimination half-life,Citation34 with eribulin predominantly metabolized by cytochrome P450 (CYP) 3A4Citation35 and eliminated principally in the feces.
Clinical trials
The unique mechanism of action of eribulin, as well as its activity in vitro and in vivo (including against paclitaxel-resistant models) and favorable pharmacokinetic properties, warranted its clinical evaluation. The National Cancer Institute started a Phase I trial in 2002, and the following year Eisai opened additional Phase I trials. Phase II trials began in 2004, followed by the first Phase III trial of eribulin in 2006.
Phase I trials and clinical pharmacokinetics
Initial clinical experience with eribulin was gained by dose escalation, initially in human trials that incorporated pharmacokinetic studies, and further pharmacokinetic data were obtained from studies in specific patient populations.
Phase I trials
Four Phase I trials have been conducted with eribulin administered intravenously as a single agent. Eribulin was stored as 1 mL vials in a 500 μg/mL solution of ethanol/water (5:95), and each vial was diluted with 4 mL of saline 0.9% to provide a 5 mL solution containing eribulin mesylate at a concentration of 100 μg/mL.
The trials used differing schedules of administration (). The National Cancer Institute study was the first to open,Citation36 followed by two Eisai-sponsored studies.Citation37,Citation38 The final Phase I study, conducted in Japan, opened later. Two studies have been published in full,Citation37,Citation38 and two only as abstracts.Citation36,Citation39
Table 1 Phase I clinical trials of eribulin mesylate
Each trial aimed to determine the maximum tolerated dose (defined as the dose recommended for Phase II studies), dose-limiting toxicities, toxicity profile, pharmacokinetics, and preliminary evidence of the antitumor activity of eribulin in patients with advanced solid tumors. Eligibility criteria were similar, ie, a cytological or histological diagnosis of advanced solid tumor, measurable or evaluable disease, age ≥18 years, life expectancy of at least 3 months, Karnofsky performance status ≥70%, and adequate liver, renal, and bone marrow reserve. The study designs successfully utilized initial accelerated dose escalation, with dose doubling and/or single patient cohorts until significant toxicity was seen, after which dose escalation and cohort sizes were conventional. Median age was about 60 years and most patients were Eastern Cooperative Oncology Group performance status 0–1. However, there were some differences with respect to tumor types, extent of prior therapy, and definition of dose-limiting toxicity.
The maximum tolerated dose of eribulin mesylate given on day 1 every 3 weeks as a 1-hour infusion was 2.0 mg/m2.Citation37 Eribulin dosed weekly on days 1, 8, and 15 every 4 weeks had an maximum tolerated dose of 1.0 mg/m2 given as a 1-hour infusionCitation38 or 1.4 mg/m2 administered over 1–2 minutes;Citation36 the maximum tolerated dose was 1.4 mg/m2 when eribulin was given over 5 minutes on days 1 and 8 every 3 weeks.Citation39 At the maximum tolerated dose, the lowest dose intensity was achieved with eribulin given as a 1-hour infusion every 3 weeks (0.66 mg/m2).Citation37 Weekly administration appeared to achieve higher dose intensities, especially with eribulin given as a bolus rather than as a 1-hour infusion (1.05 mg/m2/week and 0.75 mg/m2/week, respectively) utilizing at days 1, 8, and 15 every 4 weeks schedule or at days 1 and 8 every 3 weeks schedule (0.93 mg/m2/week).Citation36,Citation38 This suggested that maximum exposure to eribulin may be achieved with weekly dosing, given as a 2–5 minute bolus.
Across these Phase I trials, generally neutropenia and fatigue were dose-limiting, with modest levels of neuropathy reported. Treatment discontinuation was mostly due to disease progression, and there were no toxic deaths. Dose-limiting neutropenia had an earlier onset, with febrile neutropenia observed on day 7 when eribulin was given on day 1 every 3 weeks as a 1-hour infusionCitation37 than with weekly administration, where neutropenia characteristically occurred on day 15, leading to omission of treatment.Citation38 Fatigue was reported in all studies, but appeared to be less frequent and less severe with eribulin given on day 1 every 3 weeks as a 1-hour infusionCitation37,Citation39 than with administration on days 1, 8, and 15 every 4 weeks, where dose-limiting (Grades 3 and 4) fatigue was seen in four of 32 patients.Citation38 Considering that eribulin interacts with microtubules, there was relatively little neuropathy, especially with the schedule of day 1 every 3 weeks, where a single patient developed Grade 1 peripheral neuropathy at a dose two-fold greater than the ultimate maximum tolerated dose. Peripheral neuropathy appeared to be more common with weekly administration, being seen in eight of the 32 patients receiving eribulin on days 1, 8, and 15 every 4 weeks.Citation38 Any possible schedule dependence in the incidence of neuropathy should be viewed with caution because fewer patients treated with the day 1 every 3 weeks schedule had received a prior taxane. Moreover, in all cases, neuropathy was Grade 1 or 2, and led to treatment discontinuation in only one patient.Citation38 The median number of treatment cycles was two in all studies, so there were limited opportunities to identify cumulative neuropathy.
Thrombocytopenia was uncommon and mild (Grade 1 or 2), as were diarrhea and vomiting. Alopecia was also limited (Grade 1 or 2) and seen in less than one-third of patients. Other toxicities included anorexia, hypoglycemia, and hypophosphatemia.Citation36,Citation39 The Halaven prescribing information reports some prolongation of QTc on day 8 but not day 1 of treatment; likewise, there is a warning regarding potential fetal harm.
There was evidence that eribulin had clinical activity in these patients with a wide range of tumors who had exhausted established treatment options. Not surprisingly, there were no complete responses, but partial responses were seen with all four schedules. Partial responses were seen in seven patients (6.5%), four with nonsmall cell lung cancer (one unconfirmed),Citation36,Citation37,Citation39 and one each in bladder,Citation36 head and neck,Citation39 and cervical cancer (the latter also unconfirmed).Citation38 Overall, stable disease was reported in a further 38 (35.2%) patients.Citation36–Citation39
Pharmacokinetics and special populations
The clinical pharmacokinetics of eribulin were studied in the aforementioned Phase I studies and in specific pharmacokinetic trials. In all four Phase I studies, eribulin demonstrated linear, dose-proportional pharmacokinetics across the doses explored with regard to drug exposure (expressed as the area under the concentration–time curve) and maximum plasma concentrations.Citation36–Citation39 Elimination was triphasic, with an initial rapid distribution phase and extensive volume of distribution volume (approximately 100 L/m2). The terminal half-life was long (36–48 hours); at the maximum tolerated dose, plasma eribulin concentrations were above those required for cytotoxicity in vitro for prolonged periods (72 hours or more). Pharmacokinetic parameters were similar on different days of administration.
Only 5%–10% of the eribulin dose was detected unchanged in the urine, suggesting renal clearance of eribulin is of minor importance. Therefore, a specific Phase I and pharmacokinetic trial was carried out in patients with normal liver function and in those with mild-to-moderate hepatic impairment (as defined by Child–Pugh criteria).Citation40 Eribulin could be administered at 1.4 mg/m2 if hepatic function was normal, and at 1.1 mg/m2 and 0.7 mg/m2 if hepatic dysfunction was mild-to-moderate, respectively. Compared with those having normal liver function, exposure to eribulin (as reflected by the area under the concentration–time curve), increased 1.7-fold and 2.8-fold in patients with mild and moderate liver dysfunction, respectively. For patients with moderate renal impairment (creatinine clearance 30–50 mL/min) dose reduction to 1.1 mg/m2 is recommended in the Halaven prescribing information. Although eribulin is metabolized principally by hepatic CYP3A4, it appears not to cause relevant induction or inhibition of CYP3A4, so drug–drug interactions would not be expected.Citation35
A population pharmacokinetic–pharmacodynamic analysis of patients in the Phase I and II studies showed that eribulin clearance was affected by body weight, serum albumin, alkaline phosphatase, and bilirubin.Citation41 Interestingly, fatigue appeared not to be influenced by exposure to eribulin; neuropathy was, by contrast, related to cumulative eribulin exposure. The risk of Grade 4 neutropenia was correlated with both eribulin exposure and serum aspartate transaminase levels.
Phase II trials in patients with metastatic breast cancer
When the Phase II program opened in 2004, results from three of the four Phase I trials were available.Citation36–Citation38 Each schedule had shown manageable toxicity, with preliminary evidence of clinical activity, but eribulin mesylate at a dose of 1.4 mg/m2 on days 1, 8, and 15 given over 2–5 minutes every 4 weeksCitation36 was initially selected for Phase II trials, having achieved the highest dose intensity and being convenient for administration over a few minutes.
Three Phase II studies have been reported in patients with heavily pretreated advanced breast cancer. These studies were single-arm but relatively large, comprising 103, 299, and 84 patients, respectively.Citation42–Citation44 Patients had to have received both an anthracycline and a taxane, but additional treatments were also permitted; other eligibility criteria were standard, but pre-existing neuropathy Grade 1Citation42 or 1–2Citation43 was permitted. Objective response rate was the primary endpoint in all three studies, and responses were independently reviewed. To date, one trial has been reported, albeit only in abstract form.Citation44
The first Phase II breast cancer studyCitation42 initially administered eribulin as described above, but almost two-thirds of patients (44 of 70) did not receive their full cycle 1 treatment, due mainly to neutropenia on day 15. This may be because inability to administer the day 15 dose due to neutropenia had not constituted a dose-limiting toxicity in the Phase I trial.Citation36 The schedule was amended, with day 15 treatment omitted and cycles repeated every 3 weeks. The modified schedule could be delivered much more consistently in 33 subsequent patientsCitation42 and was formally evaluated in the later Phase I trial conducted in Japan.Citation39 The same modified schedule was utilized in the other two Phase II breast cancer studies.Citation43,Citation44
The activity of eribulin in all three trials is shown in . In addition to having received an anthracycline and a taxane, patients in all three trials were extensively pretreated.Citation42–Citation44 However, patients in the Japanese study were less heavily pretreated.Citation44 The objective response rate was around 10% in the two trials carried out in the West,Citation42,Citation43 and 20% in the Japanese study,Citation44 and the response duration was 4.0–5.6 months.Citation42–Citation44 The “clinical benefit rate” (defined as any partial response plus stable disease for at least 6 months) was 17%–27.5%.Citation42–Citation44 As additional secondary endpoints, progression-free survival was 2.6–3.6 months in all three studies, and overall survival was 9.0–11 months.Citation42–Citation44
Table 2 Treatment and efficacy in Phase II clinical trials on eribulin mesylate
In all three studies, eribulin had a generally good tolerability profile (). In two studies,Citation42,Citation43 routine blood counts revealed Grade 3 and 4 neutropenia in half to two-thirds of patients; however, more importantly, the incidence of febrile neutropenia was only around 5%. Between 20% and 50% of patients received granulocyte growth factors,Citation42,Citation43 indicating their widespread use as secondary prophylaxis. Grade 1 or 2 peripheral neuropathy, predominantly sensory, was seen in only one-quarter of patients despite prior taxane therapy being an eligibility requirement. Around 5% of patients experienced Grade 3 neuropathy, and there was no Grade 4 neuropathy.Citation42 In the study reported by Cortes et al,Citation43 patients with Grade 1 or 2 neuropathy at baseline were eligible. Encouragingly, although 22% of the patients treated had pre-existing neuropathy, in most cases (78%) their neuropathy did not worsen on eribulin. In the Japanese study,Citation44 myelosuppression and febrile neutropenia were substantially more common, but peripheral neuropathy was rather less common. Across all three studies, more than half of the patients reported fatigue, but this was of Grade 3 or 4 or less in 10% of patients. Other toxicities (including nausea, anemia, and anorexia) were generally mild, but alopecia (Grade 1 or 2) was reported in about half of the patients.Citation42–Citation44
Table 3 Main toxicities in Phase II clinical trials of eribulin mesylate
Phase III trials
These encouraging Phase II data led to two Phase III trials of eribulin in patients with heavily pretreated metastatic breast cancer, both of which have completed recruitment. One, the EMBRACE metastatic breast cancer trial (Study 305)Citation45 has been reported, the results of which led to approval of eribulin by the Food and Drug Administration, European Medicines Agency, and other regulatory authorities. Results from the second trial (Study 301)Citation46,Citation47 are anticipated in 2012.
The EMBRACE trial
The EMBRACE (EISAI Metastatic Breast Cancer) study evaluating the efficacy of eribulin was an open-label, multicenter, international, randomized trial with two distinctive features. Firstly, whereas most similar studies defined surrogate measures, such as objective response rates or progression-free survival, as their primary endpoints, the primary endpoint in EMBRACE was overall survival. Secondly, individual oncologists selected treatment for each woman in the control arm; this was termed “treatment of physician’s choice” (TPC). The TPC control arm was selected because at the time there was no single internationally accepted or approved chemotherapy regimen for women with heavily pretreated metastatic breast cancer. Therefore, this design had the added advantage of reflecting “real life” choices for these women. Patients in the experimental arm received eribulin mesylate at a dose 1.4 mg/m2 on days 1 and 8 every 3 weeks.
Eligible patients were over 18 years of age with measurable or evaluable locally recurrent or metastatic breast cancer. They were required to have received 2–5 prior chemotherapies, including an anthracycline and a taxane, of which at least two were to have been given in the metastatic setting. They had to have progressed within 6 months of chemotherapy, with performance status ≤2 and pre-existing neuropathy Grade ≤2. Patients were stratified according to geographical region, prior capecitabine exposure, and HER-2 status. They were randomized in a 2:1 ratio to receive either eribulin 1.4 mg/m2 on days 1 and 8 every 3 weeks or to TPC. To avoid bias, oncologists identified the potential TPC for each patient prior to randomization; TPC could be any single-agent chemotherapy, endocrine therapy, or biological therapy approved for the treatment of cancer, or best supportive care.
The primary endpoint was overall survival in the intention-to-treat population. An interim analysis was planned with the primary analysis triggered when there were 411 events. In practice, there were 422 events and 762 patients randomized by May 2009 when the study closed. Secondary endpoints were progression-free survival (by independent review), objective response rate, and safety.
In total, 135 centers in 19 different countries recruited 762 patients, of whom 508 received eribulin and 254 received TPC. As expected the two treatment arms were well balanced by stratification factors; the majority (64%) were from Western Europe, North America, or Australia, around three-quarters (73%) had received prior capecitabine, and 16% had HER-2 positive disease (20% had triple negative disease). Median age was 55 years, and patients had received a median of four prior chemotherapies; half had more than two sites of disease, and nearly two-thirds had liver metastases. No patient in the TPC arm received best supportive care alone, and only 4% received endocrine therapy. The breadth of TPC chemotherapies supported the choice of this control arm both on pragmatic grounds and also as a means of reflecting everyday clinical practice. For the 247 patients who received chemotherapy as their TPC, the most common choices were vinorelbine, gemcitabine, and capecitabine (25%, 19%, and 18%, respectively) whilst others were retreated with a taxane (n = 38) or an anthracycline (n = 24).
The study met its primary endpoint, demonstrating a statistically significant improvement in overall survival (hazard ratio = 0.81, P = 0.041). In doing so, the EMBRACE study became the first major study in more than a decade to achieve prolongation of overall survival, and eribulin was also the first single agent to demonstrate improved survival in such a heavily pretreated population. Median survival was 13.12 months for eribulin and 10.65 months for TPC, representing an improvement of just under 2.5 months (23%) for eribulin. One-year survival rates were 53.9% for eribulin and 43.7% for TPC. Planned subgroup analyses were consistent with improved overall survival in all subgroups, ie, across the geographical regions, in patients who had or had not received prior capecitabine, irrespective of estrogen/progesterone receptor or HER2 status and extent of disease. At the time of this analysis, the data were relatively immature, only 55% of patients having died. Accordingly, the regulatory authorities requested a subsequent analysis when there had been 77% of events. This unplanned analysis confirmed the findings of the primary analysis, with median survival prolonged by 2.7 months, and was statistically more robust (P = 0.014).
Secondary efficacy parameters also favored eribulin. The hazard ratio for progression-free survival by independent (0.87, 95% confidence interval [CI]: 0.71–1.05) and investigator (0.76, CI: 0.64–0.9) review was close to that for overall survival (see above), although the magnitude of improvement in progression-free survival was somewhat less than that in overall survival. The improvement in progression-free survival was significant by investigator (P = 0.002) but not by independent (P = 0.14) review; this apparent discrepancy is most likely a result of many patients, including those with nonmeasurable disease, being censored in the independent assessment of progression-free survival. Response rates were higher with eribulin than with TPC by both independent (12.2% and 4.7%, respectively; P = 0.002) and investigator (13.2% and 7.5%, respectively; P = 0.28) review; the same pattern was seen for clinical benefit rate (defined as above). Subgroup analyses of progression-free survival and relapse rate did not identify any patient group that consistently appeared not to benefit from eribulin.
Regarding the tolerability of treatment, there was no difference between the treatment arms in terms of the proportion of patients experiencing serious adverse events, fatal adverse events, or adverse events leading to dose reduction, delay, or discontinuation; rather more patients in the TPC arm had dose interruptions, most likely those receiving capecitabine. Overall, there appeared to be more toxicity in the patients receiving eribulin, but direct comparison of individual toxicities between the eribulin and TPC arms is complicated by the heterogeneous nature of the TPC treatments and their differing side effect profiles. Nevertheless, there was more myelosuppression with eribulin, the incidence of Grade 3 and 4 neutropenia being 45% and 21%, respectively. However, the majority of this neutropenia was asymptomatic and identified by routine day 15 blood counts; rates of febrile neutropenia were generally low in both arms (4.2% and 1.2%, respectively), with ≤1% fatal adverse events in both arms. Up to 20% of patients in the eribulin arm received colony-stimulating factors, suggesting widespread prophylactic (as opposed to therapeutic) use. There was more Grade 3 and 4 neuropathy, predominantly sensory, with eribulin than with TPC (8.2% and 2.0%, respectively) but this led to discontinuation of eribulin in only 4.8% of patients. This level of neuropathy can be considered relatively low for a drug targeting microtubules in a population which had all received a prior taxane, and including many patients with neuropathy at baseline. Although fatigue had been reported in earlier studies, Grade 3 and 4 fatigue was no more common in patients treated with eribulin than in those receiving TPC (8.8% and 10.1%, respectively). Alopecia was reported in a higher proportion of patients treated with eribulin than with TPC, but with many patients having pre-existing alopecia, the true incidence of hair loss with eribulin is unclear.
Strengths of the EMBRACE study include having clearly achieved for the first time prolonged overall survival that is both statistically and clinically significant in a large study of women with heavily pretreated metastatic breast cancer. The improvement in overall survival was seen across subgroups and was supported by superior progression-free survival and relapse rate. Further, the use of TPC as the control arm means the EMBRACE data reflect everyday clinical practice. One limitation, inherent in the design of the study, is that the small number of patients receiving each TPC precluded direct comparison of the efficacy or toxicity of each TPC chemotherapy with eribulin. Likewise, quality of life data were not collected; again, the large number of drugs and treatment schedules in the TPC arm would have complicated interpretation of such data. Nevertheless, prolongation of overall survival was achieved with only a modest increase in toxicity. The EMBRACE study largely underpinned the approval of eribulin by the Food and Drug Administration in November 2010 and the European Medicines Agency in March 2011 for the treatment of women with metastatic breast cancer.
Other key eribulin trials in patients with metastatic breast cancer
The second Phase III trial of eribulin in patients with advanced or metastatic breast cancer (Study 301) compared eribulin with oral capecitabine (at the approved dose of 2500 mg/m2), the latter being the only other single agent approved in those previously treated with an anthracycline and a taxane.Citation46 Study 301 differs subtlely, but significantly, from EMBRACE in other ways. Patients in Study 301 were somewhat less heavily pretreated, and had not received prior capecitabine. Randomization was 1:1, with overall survival and progression-free survival as the coprimary endpoints.Citation46 In contrast with the EMBRACE study, quality of life data were collected in Study 301, reflecting the homogeneous standard treatment arm.Citation45 Study 301 has completed recruitment of more than 1000 patients, and results are anticipated in 2012.Citation46
A Phase II study (Study 209) in which approximately 100 patients with metastatic breast cancer were randomized to receive eribulin or ixabepilone has been undertaken primarily to compare the incidence, severity, and pattern of recovery of peripheral neuropathy with these two novel tubulin-interacting agents.Citation47 An ongoing Phase I/II study (Study 203) is evaluating the tolerability and efficacy of the combination of capecitabine and eribulin.Citation48
Future directions with eribulin
Other trials of eribulin can be anticipated in patients with metastatic breast cancer, as a single agent and in combination with other cytotoxics and biologic agents, including trastuzumab. It is likely that eribulin will be evaluated in the first-line metastatic setting as well as in the neoadjuvant and adjuvant settings. To date, no specific group of patients with metastatic breast cancer has been defined which appears more or less likely to benefit from eribulin. Neoadjuvant studies may provide the best opportunity to identify factors predictive of benefit from eribulin. Preclinical work has identified the βIII subunit of tubulin as a potential biomarker. reports currently ongoing trials in patients with breast cancer registered on clinicaltrials.gov. Finally, ongoing trials are evaluating in patients with other tumor types, such as prostate cancer, nonsmall cell lung cancer, and sarcoma. Eribulin reached predefined values for progression-free survival in a recently reported Phase II study in patients with pretreated soft tissue sarcoma.Citation56
Table 4 Ongoing eribulin trials in BC registered on clinicaltrials.gov
Conclusion
Eribulin is a novel, nontaxane cytotoxic agent which targets tubulin in a specific and distinct manner. In women with heavily pretreated breast cancer, eribulin has consistently demonstrated an objective, independently confirmed response rate of 10% and a clinical benefit rate of around 20%.Citation45 However, most importantly, eribulin prolongs overall survival for women with heavily pretreated metastatic breast cancer who have previously received an anthracycline and a taxane.Citation45 This improved overall survival was in comparison with a clinically relevant control arm, ie, TPC, so reflects what can be expected in clinical practice. The improvement in survival was both statistically (P = 0.014) and clinically relevant, representing prolongation of 2.7 months in the updated analysis. The benefit from eribulin appears not to be limited to any individual clinical or molecular subgroup of women.Citation45
The tolerability of eribulin has been demonstrated in almost 1000 women treated in Phase II and III trials. Eribulin demonstrated a predictable toxicity profile, neutropenia being the most frequent toxicity (Grade 3 and 4, 54%). However, febrile neutropenia is uncommon, occurring in 5% of patients. As would be expected for a tubulin-interacting agent, peripheral neuropathy is seen, but severe neuropathy is uncommon (Grade 3–4, 8%) and is frequently managed by dose reduction or delay. In addition, eribulin is straightforward to administer, requiring no complicated premedication and being given over a few minutes.Citation45
Eribulin was approved by the Food and Drug Administration for the treatment of women with metastatic breast cancer who have previously received a taxane and an anthracycline in either the adjuvant or metastatic settings and at least two chemotherapeutic regimens for the treatment of metastatic disease. Approval has followed in Singapore in February and the European Union in March 2011. We can anticipate that eribulin mesylate at a dose 1.4 mg/m2 on days 1 and 8 every 3 weeks will become a new option and potentially the treatment of choice for these women.
Disclosure
The authors report no conflicts of interest in this work.
References
- JemalASiegelRXuJWardECancer statisticsCA Cancer J Clin20106027730020610543
- ParkinDMFernándezLMUse of statistics to assess the global burden of breast cancerBreast J200612Suppl 1S70S8016430400
- GiordanoSHBuzdarAUSmithTLKauSWYangYHortobagyiGNIs breast cancer survival improving?Cancer2004100445214692023
- KenneckeHYerushalmiRWoodsRMetastatic behavior of breast cancer subtypesJ Clin Oncol2010283271327720498394
- GianniLNortonLWolmarkNSuterTMBonadonnaGHortobagyiGNRole of anthracyclines in the treatment of early breast cancerJ Clin Oncol2009274798480819687331
- MartínMSeguíMAAntónAAdjuvant docetaxel for high-risk, node-negative breast cancerN Engl J Med20103632200221021121833
- LoeschDGrecoFASenzerNNPhase III multicenter trial of doxorubicin plus cyclophosphamide followed by paclitaxel compared with doxorubicin plus paclitaxel followed by weekly paclitaxel as adjuvant therapy for women with high-risk breast cancerJ Clin Oncol2010282958296520479419
- BedardPLDi LeoAPiccart-GebhartMJTaxanes: Optimizing adjuvant chemotherapy for early-stage breast cancerNat Rev Clin Oncol20107223619997076
- O’ShaughnessyJExtending survival with chemotherapy in metastatic breast cancerOncologist200510Suppl 3202916368868
- PerezEALerzoGPivotXEfficacy and safety of ixabepilone (BMS-247550) in a Phase II study of patients with advanced breast cancer resistant to an anthracycline, a taxane, and capecitabineJ Clin Oncol2007253407341417606974
- SeoHYLeeHJWooOHPhase II study of vinorelbine monotherapy in anthracycline and taxane pre-treated metastatic breast cancerInvest New Drugs20112936036519943080
- SpielmannMLlombart-CussacAKallaSSingle-agent gemcitabine is active in previously treated metastatic breast cancerOncology20016030330711408796
- RobertNJConklingPRO’RourkeMAResults of a Phase II study of pemetrexed as first-line chemotherapy in patients with advanced or metastatic breast cancerBreast Cancer Res Treat201112610110821188632
- ChewHKDoroshowJHFrankelPPhase II studies of gemcitabine and cisplatin in heavily and minimally pretreated metastatic breast cancerJ Clin Oncol2009272163216919307510
- MurphyCGSeidmanADEvolving approaches to metastatic breast cancer previously treated with anthracyclines and taxanesClin Breast Cancer20099S58S6519596644
- NewmanDJCraggGMBattershillCNTherapeutic agents from the sea: Biodiversity, chemo-evolutionary insight and advances to the end of Darwin’s 200th yearDiving Hyperb Med20093921622522752743
- HirataYUemuraDHalichondrins: Antitumor polyether macrolides from a marine spongePure Appl Chem198658701710
- TowleMJSalvatoKABudrowJIn vitro and in vivo anticancer activities of synthetic macrocyclic ketone analogues of halichondrin BCancer Res2001611013102111221827
- PettitGRHeraldCLBoydMRIsolation and structure of the cell growth inhibitory constituents from the western Pacific marine sponge Axinella spJ Med Chem199134333933401956053
- AicherTDBuszekKRFangFGTotal synthesis of halichondrin B and Norhalichondrin BJ Am Chem Soc199211431623164
- StamosDPChenSSKishiYNew synthetic route to the C.14-C.38 segment of halichondrinsJ Org Chem19976275527553
- WangYHabgoodGJChristWJKishiYLittlefieldBAMelvinJYStructure-activity relationships of halichondrin B analogues: Modifications at C.30+/−C.38Bioorg Med Chem Lett2000101029103210843209
- BaiRLPaullKDHeraldCLMalspeisLPettitGRHamelEHalichondrin B and homohalichondrin B, marine natural products binding in the vinca domain of tubulin. Discovery of tubulin-based mechanism of action by analysis of differential cytotoxicity dataJ Biol Chem199126615882158891874739
- JordanMAKamathKMannaTThe primary antimitotic mechanism of action of the synthetic halichondrin E7389 is suppression of microtubule growthMol Cancer Ther200541086109516020666
- DabydeenDABurnettJCBaiRComparison of the activities of the truncated halichondrin B analog NSC 707389 (E7389) with those of the parent compound and a proposed binding site on tubulinMol Pharmacol2006701866187516940412
- JordanMAWilsonLMicrotubules as a target for anticancer drugsNat Rev Cancer2004425326515057285
- AldayPHCorreiaJJMacromolecular interaction of halichondrin B analogues eribulin (E7389) and ER-076349 with tubulin by analytical ultracentrifugationBiochemistry2009487927793819586046
- OkounevaTAzarenkoOWilsonLLittlefieldBAJordanMAInhibition of centromere dynamics by eribulin (E7389) during mitotic metaphaseMol Cancer Ther200872003201118645010
- KuznetsovGTowleMJChengHInduction of morphological and biochemical apoptosis following prolonged mitotic blockage by halichondrin B macrocyclic ketone analog E7389Cancer Res2004645760576615313917
- KuznetsovGTenDykeKYuMJLittlefieldBAYuJLittlefieldBAAntiproliferative effects of halichondrin B analog eribulin mesylate (E7389) against paclitaxel-resistant human cancer cells in vitroProceedings of the annual meeting of American Association for Cancer ResearchLos Angeles, CAApril 14–18, 2007 Abstr C58
- AgoulnikSKuznetsovGTendykeKSensitivity to halichondrin analog E7389 and hemiasterlin analog E7974 correlates with βIII tubulin istotype expression in human breast cancer cell linesJ Clin Oncol20052316 Suppl201215774792
- AlleyMCSmithACDonohueSJComparison of the relative efficacies and toxicities of halichondrin B analoguesProceedings of the 17th American Association for Cancer Research-National Cancer Institute-European Organization for Research on Cancer International ConferenceNovember 14–18, 2005 Abstr C230
- SmithJAWilsonLAzarenkoOEribulin binds at microtubule ends to a single site on tubulin to suppress dynamic instabilityBiochemistry2010491331133720030375
- NewmanSEribulin, a simplified ketone analog of the tubulin inhibitor halichondrin B, for the potential treatment of cancerCurr Opin Investig Drugs2007810571066
- ZhangZYKingMPelletierRDWongYNDelineation of the interactions between the chemotherapeutic agent eribulin mesylate (E7389) and human CYP3A4Cancer Chemother Pharmacol20086270771618431572
- SynoldTWMorganRJNewmanEMA Phase I pharmacokinetic and target validation study of the novel anti-tubulin agent E7389: A California Cancer Consortium trialJ Clin Oncol20052316 Suppl3036
- TanARRubinEHWaltonDCPhase I study of eribulin mesylate administered once every 21 days in patients with advanced solid tumorsClin Cancer Res2009154213421919509146
- GoelSMitaACMitaMA Phase I study of eribulin mesylate (E7389), a mechanistically novel inhibitor of microtubule dynamics, in patients with advanced solid malignanciesClin Cancer Res2009154207421219509177
- MinamiHMukoharaTNagaiSMukaiHNamikiMA Phase 1study of eribulin mesylate (E7389) in patients with refractory cancersProceeding of 20th American Association for Cancer Research-National Cancer Institute-European Organization for Research on Cancer Symposium on Molecular Targets and Cancer TherapeuticsGeneva, SwitzerlandOctober 21–24, 2008 Abstr 446
- WitteveenPMarchettiSMergui-RoelvinkMEribulin mesylate pharmacokinetics in patients with hepatic impairmentJ Clin Oncol2010282582
- GuptaAMouldDRReydermanLPopulation pharmacokinetic-pharmacodynamic (PK/PD) analysis of safety and efficacy of eribulin in metastatic or locally advanced breast cancer patientsProceedings of European Society of Medical Oncology annual meetingStockholm, SwedenSeptember 12–16, 2008 Abstr 446
- VahdatLTPruittBFabianCJPhase II study of eribulin mesylate, a halichondrin B analog, in patients with metastatic breast cancer previously treated with an anthracycline and a taxaneJ Clin Oncol2009272954296119349550
- CortesJVahdatLBlumJLPhase II study of the halichondrin B analog eribulin mesylate in patients with locally advanced or metastatic breast cancer previously treated with an anthracycline, a taxane, and capecitabineJ Clin Oncol2010283922392820679609
- IwataHAogiKMasudaNEfficacy and safety of eribulin in Japanese patients with advanced breast cancerJ Clin Oncol2010281081
- CortesJO’ShaughnessyJLoeschDA Phase III open-label randomised study (EMBRACE) of eribulin monotherapy versus treatment of physician’s choice in patients with metastatic breast cancerLancet201137791492321376385
- ClinicalTrials.govE7389 versus capecitabine in patients with locally advanced or metastatic breast cancer previously treated with anthracyclines and taxanes and refractory to the most recent chemotherapy Available at: http://clinicaltrials.gov/show/nct00337103Accessed May 20, 2011
- TwelvesCCortesJVahdatLTWandersJAkereleCKaufmanPAPhase III trials of eribulin mesylate (E7389) in extensively pretreated patients with locally recurrent or metastatic breast cancerClin Breast Cancer20101016016320299316
- ClinicalTrials.govA study comparing eribulin mesylate and ixabepilone in causing or exacerbating neuropathy in patients with advanced breast cancer Available at: www.clinicaltrials.govAccessed May 20, 2011
- ClinicalTrials.govA Phase 1b/2, multicenter, randomized, open-label, dose-escalation and confirmation study of eribulin in combination with capecitabine Available at: www.clinicaltrials.govAccessed May 20, 2011
- ClinicalTrials.govDose dense doxorubucin and cyclophosphamide followed by eribulin mesylate for the adjuvant treatment of early stage breast cancer Available at: www.clinicaltrials.govAccessed May 20, 2011
- ClinicalTrials.govA study of single-agent eribulin mesylate as first-line therapy for locally recurrent or metastatic human epidermal growth factor receptor two (HER2) negative breast cancer Available at: www.clinicaltrials.govAccessed May 20, 2011
- ClinicalTrials.govEribulin with trastuzumab as first-line therapy for locally recurrent or metastatic HER2 positive breast cancer Available at: www.clinicaltrials.govAccessed May 20, 2011
- ClinicalTrials.govA study of CDX-011 (CR011-vcMMAE) in patients with advanced GPNMB-expressing breast cancer Available at: www.clinicaltrials.govAccessed May 20, 2011
- ClinicalTrials.govCompassionate use of eribulin for the treatment of advanced breast cancer refractory to all other marketed therapies Available at: www.clinicaltrials.govAccessed May 20, 2011
- ClinicalTrials.govAn open-label, multi-center, expanded access program with eribulin for the treatment of advanced breast cancer refractory Available at: www.clinicaltrials.govAccessed May 20, 2011
- SchoffskiPRay-CoquardILCioffiAActivity of eribulin mesylate (E7389) in patients with soft tissue sarcoma (STS): Phase II studies of the European Organisation for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group (EORTC 62052)J Clin Oncol20102810031