Abstract
The discovery that breast cancers contain stem-like cells has fuelled exciting research in the last few years. These cells are referred to as breast cancer stem cells (BCSCs) and are thought to be involved in tumor initiation, progression, and metastasis. Being intrinsically resistant to chemo- and radiotherapy, they are also considered responsible for recurrence of the disease after treatment. BCSCs have been suggested to be at the basis of tumor complexity, as they have the ability to self-renew and give rise to highly proliferating and terminally differentiated cancer cells that comprise the heterogeneous bulk of the tumor. There has been much speculation on the BCSC model, and in this review we address some fundamental questions, such as the identity of BCSCs and their involvement in tumor intra- and interheterogeneity. As an alternative to the BCSC model, we discuss clonal evolution, as both theories show extensive evidence in support of their arguments. Finally, we discuss a unifying idea that reconciles both models, which is based on stem cell plasticity and epigenetic modifications induced by the tumor microenvironment. The implications of cancer stem cell plasticity for drug discovery and future therapeutic interventions are presented.
Breast cancer: a complex and heterogeneous disease
Breast cancer is one of the most common causes of cancer-related mortality in women worldwide (following lung cancer), with more than one million women diagnosed every year, and half a million dying from this disease. Although medical advances have contributed to early detection and better treatment, the mortality rate of women with breast cancer is still relatively high due to recurrence and metastasis.Citation1 Breast cancer represents a major clinical challenge as it is a complex disease, and presents with significant variability in tissue histopathology, metastatic behavior, response to treatment, and patient outcomes. At a cellular level, breast cancer is regarded as a heterogeneous disease. This heterogeneity profoundly impacts treatment, as combinatorial therapies are required to target different cancer cells. Heterogeneity is not only a feature of different breast tumor subtypes (interheterogeneity) but also of the same tumor (intraheterogeneity).Citation2 In terms of interheterogeneity, different breast cancer subtypes can be classified based on clinical and histological factors, which include tumor grade, size, stage, and lymph node metastasis. According to the World Health Organization, there are at least 18 different histological subtypes of breast cancer.Citation3 Although these histological and clinical subsets give a detailed account of different tumors, there is still variability with grading and diagnosis.
Molecular profiling of tumors shows that breast cancer is a heterogeneous set of different diseases, determined by various molecular alterations, rather than being a single disease with multiple manifestations.Citation4 Genetic and epigenetic insults contribute to breast carcinogenesis.Citation5 These alterations cause aberrant expression of oncogenes and silencing of tumor suppressor genes with consequent disruption in gene networks regulating normal tissue homeostasis, such as cell proliferation, differentiation, motility, apoptosis, and growth.Citation6 Gene expression profiling performed across breast cancer subsets identified estrogen receptor alpha (ER) positive (ER+) and negative (ER−) tumors as two distinct cancer types. Furthermore, it allowed further subclassification across five molecular subtypes depending on their “intrinsic gene expression” signature. These include normal-like, luminal A, luminal B, HER2 positive (HER2+), and basal-like subtypes.Citation7–Citation9 Luminal A tumors are defined by the expression of both ER+ and/or progesterone receptors (PR+/PR−), and by the absence of HER2 amplification. Luminal B tumors are similar to luminal A, but include the amplification of HER2. HER2+ tumors are defined by HER2 expression and may lack the expression of ER and PR. Basal-like tumors are defined by the absence of ER, PR, and HER2 expressions and are further subdivided into basal A and basal B.Citation7,Citation10 More recently, a molecular signature identified as ‘claudin-low’ has been found to overlap with the basal B subtype. Claudin-low tumors lack the expression of ER, PR, and HER2, and are therefore also identified as “triple negative.” Normal-like breast cancer shows a gene signature similar to that of normal breast tissue.Citation11 Less common than other subtypes, normallike cancers are of an ambiguous origin, and it is still debated whether they may represent breast tissue containing too few detectable cancer cells at the time of analysis.Citation12
Breast cancer subtypes exhibit differences in the incidence of the disease, survival rates, and response to treatment. Luminal tumors (almost exclusively ER+) are the most common, and are associated with positive outcomes as they are treatable with hormonal therapy (tamoxifen). Luminal A subtypes are less proliferative than luminal B, and therefore have a better outcome. HER2+ tumors are highly proliferative, and present with worse outcomes even if treated with the anti-HER2 antibody trastuzumab (Herceptin®; Genentech USA, Inc, San Francisco, CA). Basal-like breast cancers represent high grade cancers with poor patient outcomes. Although somewhat sensitive to chemotherapy, these cancers are associated with high levels of recurrence after treatment. Targeted therapies, with the exception of poly (ADP-ribose) polymerase (PARP) inhibitors, are lacking for basal-like breast cancers ().Citation13 New research has revealed that the complexity of breast cancer is higher than previously expected. Indeed, a screening of 2000 breast tumors that combined inherited and acquired genetic alterations with gene expression data, highlighted a novel molecular stratification of tumors with ten different subtypes.Citation14
The cellular origin of different breast tumor subtypes is still unclear. Some breast cancer subtypes have similar genetic and molecular compositions as their normal mammary cell counterparts. For instance, luminal subtypes have a similar molecular makeup to luminal mature non-clonogenic cells in that they are both ER+ and PR+. They also express characteristic luminal markers, such as CK18, CK19, CD24, MUC1, and ESA. In contrast, the basal B/claudin-low subtypes lack the ER, PR, and HER2 expression and express markers characteristic of the basal lineage, such as CK14, CD49f, and CD44.Citation15 This evidence would suggest that different cancer subtypes originate from either luminal or basal/myoepithelial progenitors within the normal tissue. However, this correlation does not exist when basal-like/BRCA1 mutant tumors are considered, as they seem to originate from luminal progenitor cells, rather than from basal progenitors.Citation16,Citation17 The origin of all tumor subtypes – whether it is a cell with multilineage potential, a progenitor cell, or even a differentiated cell – is still under debate. Moreover, this quest is further complicated by the occurrence of intratumor heterogeneity within breast cancer subtypes. Intraheterogeneity is typically observed at the histological level, with different cell types and/or morphological appearances making up the bulk of the tumor. This is reflected in variable gene expression signatures and is best exemplified by the variable ER expression within a patient tumor.Citation18 Two different models, supported by experimental findings, explain the origin of tumor heterogeneity: stem cell hierarchy and clonal evolution.
Breast cancer stem cells
The idea that cancer is driven by cells with stem cell-like features is not new, but has received renewed interest in recent years. The theory that cancer arises from stem cells was developed in the late nineteenth century when a correlation between embryonic stem cells and cancer was established among teratocarcinomas.Citation19 The existence of malignant stem cells, also known as cancer stem cells (CSCs), was first discovered in acute myeloid leukemia; since then, CSCs have been identified and isolated in many solid tumors including breast, prostate, brain, and lung.Citation20 According to the CSC hypothesis, the tumor is organized into aberrant hierarchies in which the CSC lies at the apex, and highly proliferating progenitors and terminally differentiated cancer cells reside at the bottom. In this model, CSCs would sustain tumor growth by symmetrical and asymmetrical self-renewal, whereas lineage-committed progenitor and differentiated cells would make up the heterogeneous bulk of the tumor ().
The CSC hierarchy model predicts that CSCs originate from the transformation of normal stem cells and, in this context, they are believed to be a rare population of cells more tumorigenic than the non-CSC population.Citation20 However, the origin of CSC is at the center of controversy, as they may not necessarily derive from transformed stem cells, as the name implies. Therefore, many researchers prefer to refer to these cells as ‘cancer-initiating cells’ or ‘cancer-propagating cells.’ For the simplicity of semantics, we shall refer to them as CSCs in this review.Citation21 The cell-of-origin of breast CSCs (BCSCs) is not yet known, but there are two possibilities: they either originate from undifferentiated mammary stem cells (MaSCs) or committed stem/progenitor cells through genetic and epigenetic reprogramming.Citation21
The mammary gland is a very dynamic tissue and it is organized in a hierarchical fashion where MaSCs give rise to highly proliferating progenitors and differentiated cells of the epithelial and myoepithelial lineages (). Most of our knowledge about MaSCs comes from the mouse model, where experiments of serial transplantation into the mammary fat pad show that single basal stem cells, identified as CD49f+/CD29+/CD24low repopulating cells, can give rise to different mammary structural units.Citation22,Citation23 According to the BCSC theory, it is thought that long-living MaSCs might represent a likely target for malignant transformation; however, recent lineage-tracing experiments and clonal analyses have defined the hierarchical structure of the mouse mammary gland and identified both luminal and myoepithelial long-lived unipotent stem/progenitor cells as being able to clonally expand and maintain proliferation in adulthood.Citation24 Therefore, these cells could also be a target for cellular transformation. Less is known about the organization of the human mammary gland and most of the data are inferred from experiments that combine flow cytometry, in vitro assays, and xenotransplantation. Human MaSCs have been shown to have a CD49f+/ESA−/low phenotype, suggesting a basal location of these cells in the gland;Citation16,Citation25 however, the precise structure of the human MaSCs hierarchy is still not fully understood. For instance, it is unclear whether MaSCs differentiate into a common bipotent progenitor that in turn gives rise to committed progenitors, especially since specific markers for such early lineages are currently lacking.Citation26 Indeed, different studies have identified bipotent progenitor cells as luminal ESA+/CD49f+/MUC1− or basal CD49f+/ESA−/low cell populations, suggesting that two different stem/progenitor cell populations could also exist in the human breast.Citation26,Citation27
BCSCs were first identified and isolated by the virtue of cell-surface expression markers, CD44 and ESA, and the absence of CD24.Citation28 Cells identified as Lin−/ESA+/CD44+/CD24−/low were found to be more tumorigenic compared to the CD44+/CD24+/ESA− cell population, and they were also able to generate tumors in non-obese diabetic/severe combine immunodeficient mice. Importantly, the transplanted tumors recapitulated the same heterogeneity of the original tumor, even after serial transplantation. The molecular characterization of CD44+ and CD24+ cells also confirmed that CD44+ cells express basal stem cell markers, while CD24+ cells express markers characteristic of differentiated luminal cells.Citation29 However, only a fraction of the CD44+/CD24−/low cells are highly tumorigenic, indicating that expression of these markers can be used to enrich BCSCs, but they may not identify a pure CSC population.Citation30,Citation31 In response, other breast cancer stem cell markers have been investigated. ALDH1 has been shown to be a BCSC marker, and cancer cells that have a CD44+/CD24−/low/ALDH1high profile are more tumorigenic, with as few as 20 cells being able to generate tumors after transplantation.Citation32 However, ALDH1 activity has been shown to be low or absent in normal mammary stem cells, but high in luminal progenitor cells, again questioning the true identity of BCSCs.Citation33
BCSCs can also be enriched and cultured in non-adherent conditions through their ability to grow in suspension, bypassing anoikis and forming ‘mammospheres’.Citation34 The generation of mammospheres can be used to measure the proliferative and the self-renewal abilities of BCSCs at clonal density, with stem and early progenitor cells forming a greater number of mammospheres compared to committed cells over the course of several generations. Importantly, mammosphere cultures enriched with undifferentiated cells demonstrated increased tumor-initiating capacity in vivo.Citation30,Citation35 Mammospheres comprise a heterogeneous cell population and it is not known whether clonal sphere-forming cells represent stem/progenitor cells with basal characteristics, luminal characteristics, or both. An elegant study conducted by Pece et al identified normal mammary sphere-forming cells as quiescent MaSCs (which are high retainers of the PKH26 tracking dye), and these were found to be expressing CD24, DLL, DNER, and CD49f.Citation36 PKH26high stem cells self-renew asymmetrically giving rise to luminal or myoepithelial progenies. Importantly, cells with this molecular signature were found to be particularly enriched in poorly differentiated breast cancers.Citation36 The frequency of BCSCs, characterized by the described markers, depends on the tumor subtype and histological grade, with high-grade tumors being the most enriched.Citation37
It is now becoming evident that different breast cancer subtypes may have different cellular origins (). Several studies have shown that primitive basal MaSCs are the likely cell of origin of basal B/claudin low and metaplastic cancers, whereas luminal progenitors can generate luminal and basal-like tumors. Indeed, transformation of luminal ESA+ progenitor cells by oncogene overexpression can give rise to both ER+ (luminal) and ER− (basal-like and possibly HER2+) cancers.Citation12,Citation15,Citation16,Citation38 Consistent with this notion are recent findings showing that luminal progenitors are the cell of origin for BRCA1 and TP53 mutated basal-like breast cancers.Citation16,Citation17,Citation39
Controversies of the BCSC model
To date, in vivo xenograft and in vitro differentiation data suggest that the human mammary gland is organized in a hierarchical fashion, supporting the BCSC hypothesis.Citation40 However, the molecular identity of progenitor cells remains elusive, and it is therefore uncertain whether BCSCs represent transformed MaSCs, progenitor cells, or both. Recent lineage tracing experiments have shown how mouse skin, intestine, and brain CSCs initiate and sustain tumors in their own environment.Citation27,Citation38,Citation41 These studies, which elegantly demonstrate CSC activity in intact tumors, offer much promise for the CSC debate. Similar experiments conducted with BCSCs would greatly benefit our understanding of breast cancer even if the recapitulation of the human disease in mice remains a limitation.Citation42 At present, transplantation into humanized mammary fat pads of immunocompromised mice is the best available assay for testing human BCSC function. It is thought that BCSCs are highly tumorigenic, with only a small number of cells required to form tumors when compared to non-stem cells.Citation20
This assumption has been a subject of criticism. First, the tumorigenic behavior of cancer cells may vary probabilistically and, given optimal conditions, any tumor cell may have the same probability of exhibiting tumorigenic behavior.Citation43 Human tumor cells are not easily conducive to engraftment due to differences in the microenvironment of the mouse mammary fat pad.Citation44 A second argument refers to the level of immunosuppression in some mouse models.Citation45 A study by Quintana et al examining melanoma showed that the tumorigenicity of cancer cells can be dramatically increased by transplantation into non-obese diabetic/severe combine immunodeficient gamma mice, which is a mouse strain with superior immunodeficiency.Citation46 This study suggests that putative BSCSs may be more tumorigenic simply because of preferential or improved engraftment ability.Citation41
Since the initial publication by Al-Hajj et al, which identified BCSCs as a Lin-/ESA+/CD44+/CD24−/low (very similar to the CD49f+/ESA−/low/MUC1− phenotype), other studies have followed and they have identified BCSCs as being from the CD44+/CD24−/low population.Citation28,Citation29,Citation47 However, the observation that luminal tumors contain a minimal or non-existent CD44+/CD24− cell population, and the finding that CD44− cells are also tumorigenic in serial dilution transplantation, questions the true identity of BCSCs and creates doubt in that CD44+ cells may simply represent cells with better engraftment potential.Citation31 Indeed, a degree of developmental plasticity has been observed in the BCSC hierarchy, whereby CD44+/CD24+ and CD44+/CD24− cells can interconvert into one another and can generate tumors after xenotransplantation.Citation48 Interestingly, the CD44+ cell signature is associated with a high risk of distant metastasis, even if metastatic lesions are enriched with luminal CD24+ cells.Citation29 This indicates a phenotypic switch during tumor progression that is independent of the hierarchical differentiation program. This idea is consistent with the clinical observation that CD44+/CD24− cells are not correlated with breast cancer progression or prognosis, but favor distant metastasis.Citation29 Therefore, the notion that the CD44+/CD24− phenotype represents a universal BCSCs profile is somewhat simplistic, although basal cells identified by this profile seem to be the cell-of-origin of claudin-low/basal B breast cancer.Citation16
Another assumption of the stem cell hierarchy model is that BCSCs are a rare population of cells, but this may not necessarily be the case. Indeed, claudin-low, basal-like, and HER2+ cancers are highly enriched for BCSCs, and this characteristic is also retained in cancer cell lines derived from their respective primary tumors.Citation12 The abundance of BCSCs is regulated by self-renewal mechanisms which are dependent on the function of the tumor suppressor gene TP53. Mutated or attenuated TP53 signaling confers symmetrical self-renewal, whereby cells can give rise to two identical BCSCs at each round of cell division. Under these conditions, the MaSC differentiation process is compromised and shifted towards an accumulation of undifferentiated BCSCs. Cell tracking experiments have proven symmetrical self-renewal in mammospheres generated from HER2+ transgenic mouse tumors and human basal cancer cell lines (personal observation).Citation49 Altogether, this evidence casts some doubt on the BCSC hierarchy model, and it may explain why the BCSC hypothesis is not universally accepted by the scientific and medical communities. The key challenge of the BCSC model is the identification of the “primitive malignant stem cell” at the origin of different breast cancers, and recent evidence suggests that this model will need to evolve to accommodate stochastic events that contribute to inter- and intratumor phenotypic and genotypic heterogeneity. As an alternative, tumor heterogeneity can be explained by the clonal evolution model.
An alternative model: clonal evolution
In contrast to the stem cell hierarchy theory, the clonal evolution model proposes that different clones of cancer cells arise with different selection pressures and microenvironment influences, which can include endogenous and exogenous factors.Citation50 Clones of transformed cells can accumulate when cellular hyperproliferation is combined with genetic instability. In this way, tumor heterogeneity is caused by noise-driven gene expression differences, as well as the growth of transformed cells that do not necessarily involve stem cells.Citation51 Tumorigenesis is the result of a collection of random mutations that are associated with the appearance of dominant cell clones with growth advantages resulting from activated oncogenes and/or inactivated tumor suppressor genes that are selected by a Darwinian process.Citation52 The transformation of cells through genetic mutations is a stochastic process, whereby the phenotypic change in a cell is not predetermined; rather, the different cell clones are generated by random mutation hits. Through Darwinian selection, clones with advantageous mutations are selected to be dominant within the tissue, whereas disadvantageous mutations are discarded. The neutral clones are retained within the population causing a genetic drift. As a consequence, a selection of clones produces a dynamic state during cancer progression: some clones have no desirable mutations for further survival, while others have a selective advantage. Therefore, different parts of the tumor could be undergoing different selective pressure, owing to heterogeneity ().Citation53
When considering clonal heterogeneity, there is evidence that supports a close clonal relationship between the primary and metastatic tumors. However, in some tumors, metastatic spread occurs at the early stages of tumor evolution, hence the primary and metastatic tumors may evolve to have distinct genetic identities over time.Citation53 The first report that investigated clonal diversity of breast tumors at the single cell level was published in 2011.Citation54 This study reported the existence of punctuated clonal expansion with few persistent intermediates during tumor progression, rather than a gradual expansion of tumorigenic cells. A similar observation was recently reported in a study of next generation sequencing of 104 primary triple-negative breast cancer cases.Citation55 At the time of diagnosis, these tumors displayed a wide spectrum of mutation heterogeneity and clonal evolution. Mutations in the TP53, PIK3CA, and PTEN genes seemed dominant compared to other genetic defects, but they were sometimes present at such low frequencies that they did not appear to be cancer founder mutations. Therefore, mutational heterogeneity is present at the onset of triple negative breast cancer, and patients present with either low-clonality or high-clonality cancers.
Clonal evolution can also explain the phenomenon of intertumor heterogeneity observed in different cancer subtypes. One study that analyzed the contribution of germline and somatic alterations in a cohort of 2000 breast cancers revealed considerable tumor heterogeneity, and highlighted novel subtypes occurring with different frequencies in the population.Citation14 However, genetic heterogeneity may not solely explain the phenotypic diversity of tumor cancer cells, as cancer cell behavior can also be influenced by the environment, which can alter gene expression by epigenetic modifications. The microenvironment is not homogenous in a tumor, as different regions within the tissue have varying densities of vasculature, different numbers and types of immune cells, and varying compositions of the extracellular matrix. Again, on its own, the clonal evolution model may not fully explain the complexity of tumor heterogeneity as it needs to take in to account non-genetic (epigenetic) influences on hereditable phenotypes.
Stem cell plasticity and cancer: a unifying idea
It is becoming widely accepted that the BCSC and clonal evolution models are not mutually exclusive; they both contribute to the explanation of tumor heterogeneity. A unifying idea is presented that discusses the role of the inherent developmental plasticity of stem cells. Stochastic events that affect stem cell function – either genetic alterations or epigenetic modifications induced by the tumor microenvironment – can induce cellular transformation and confer cancer cells with stem cell-like characteristics. Although cell-lineage restriction programs are established during embryonic development, adult stem cells maintain a degree of plasticity which is necessary for tissue repair and/or turnover.Citation19 This flexibility is maintained by reversible epigenetic modifications which regulate gene expression in a cell-specific manner. Epigenetic modification of the chromatin regulates gene expression without changing the DNA sequence. This is accomplished via DNA methylation, modification of histone tails, and modulation by non-coding RNAs such as microRNA (miRNA).Citation56 With changes in chromatin conformation, epigenetic modifications establish heritable transcriptional states responsible for the maintenance of cell identity and function. Epigenetic alterations are observed at the early stages of carcinogenesis, and they play a critical role in tumor initiation and CSC plasticity. Normal stem cells are vulnerable to epigenetic defects when induced to sustained self-renewal, resulting in silencing of tumor suppressor genes.Citation57,Citation58
Many tumor suppressor genes are developmentally regulated-genes that regulate the fate of stem cells. Their epigenetic silencing can generate CSCs locked in a self-renewal state with impaired differentiation potential; indeed, several studies have shown that tumor suppressor genes are more likely to become silenced by DNA methylation in cancer.Citation59–Citation61 Reprogramming experiments have demonstrated that epigenetic landscapes are plastic and that they can be influenced and manipulated to change cell fate. The ability of stem cells, progenitor cells, or differentiated cells to transform into CSCs shows the intrinsic plasticity of these cells.Citation19 Cells can acquire several rounds of carcinogenic insults before transformation, with progeny being susceptible to further insults, resulting in genetically and sometimes phenotypically different cancer cells.Citation20 Since cellular and molecular phenotypes can be determined by genetic and epigenetic alterations affecting differentiation programs, it can be problematic to trace the cell-of-origin of different cancer types. Therefore, it is paramount to stress that the similarities of the genetic signatures between normal MaSCs and BCSCs do not necessarily reflect their direct association during transformation. For instance, BRCA1 and TP53 mutations can affect the differentiation potential of luminal progenitor cells, leading to a basal tumor phenotype.Citation16,Citation17,Citation39
Phenotypic switches in response to stochastic events, is one of the characteristics of CSCs. These involve the coexistence of different genetic and epigenetic states during cancer progression.Citation19 BCSCs can shift between a stem cell and a non-stem cell state, owing to its plasticity. This was first identified in breast cancer cell lines where non-stem cells (CD44+/CD24+) were able to generate CD44+/CD24− BCSCs with tumorigenic properties and vice versa, depending upon activation of the Activin/Nodal pathway.Citation48 One prediction of the stochastic and cell plasticity model is that the proportion of cell populations in a given tumor is determined by a phenotypic equilibrium reached over time. Using Markov’s mathematical model, it has been shown that the change in the state of a cancerous cell is not predetermined by its previous cellular memory, but by its ability to maintaining equilibrium to reach a stable state. This is exemplified in breast cancer cell lines whereby BCSCs, luminal, and basal cells isolated according to the expression of a panel of cell surface markers, can change their phenotype over time into a metastable state. This metastable state is characterized by the same phenotype of the parental cell line, with luminal and basal committed cells each giving rise to a similar proportion of BCSCs of the parental cell line.Citation62 Therefore, selective pressure could cause an interconversion of these cellular states, so that a metastable cell state is generated and dominates the tumor population. Consistent with this model, BCSCs could arise from more differentiated cells, following a bidirectional interconversion along the stem cell hierarchy.Citation63 Based on this notion it is easy to reconcile how selective mutations that confer cancer cells with self-renewal ability can create dominant clones with BCSC characteristics during tumor progression. Different dominant clones could harbor distinct genetic alterations, and these could be selected independently under selective pressure ().
Selective pressure can also take control of normal developmental processes and affect CSC plasticity.Citation64 Epithelial-to-mesenchymal transition (EMT) a reversible embryonic program that allows for a transition between cellular phenotypes during gastrulation, is recapitulated during tumor progression and metastasis when cells change from an epithelial to a motile mesenchymal phenotype. Motile cancer cells can therefore invade neighboring and distant tissues and then colonize new sites after undergoing a reverse mesenchymal-to-epithelial transition (MET).Citation20 This interconversion explains the previously described plasticity of CD44+/CD24− cells, and the enrichment for CD24+ cells at the site of metastasis as cells undergo MET. EMT, induced by tumor microenvironment signals (TGFβ, Notch, EGF, Hedgehog, Wnt) induces BCSC properties, such as self-renewal and metastatic ability, in non-tumorigenic cells.Citation65,Citation66 TGFβ and other cytokines produced by the tumor microenvironment are directly involved in the epigenetic regulation of EMT, as well as in the acquisition of the BCSC phenotype. This effect is mediated by DNA methylation alterations that can cause silencing of adhesion molecules (eg, hypermethylation of CDH1 or E-Cadherin) and/or activation of EMT inducers by DNA hypomethylation.Citation66 DNA methylation analysis of CD44+/CD24− cells isolated from neoplastic breast tissue shows hypomethylation of several transcription factors involved in EMT, including the transcription factor FOXC1.Citation67 TGFβ can also induce epigenetic silencing of miR-200a, a key microRNA (miRNA) involved in the regulation of EMT by inducing over-expression of the histone deacetylase SIRT1 and DNA methylation at the gene promoter region.
Two miRNA subfamilies, miR-200c/141 and miR-200a/200b/429, are involved in the negative regulation of EMT, as they target EMT-inducing transcription factors ZEB1 and ZEB2.Citation68 Both miR-200 gene cluster promoters are frequently hypermethylated, and the epigenetic silencing of miR-200c has been found in BCSCs.Citation69 In addition, silencing of miRNAs negatively affects stem cell differentiation and induces self-renewal. This has been observed for miR200c given that epigenetic silencing is associated with sustained expression of the stem cell self-renewal-regulating factors KLF4 and BMI1.Citation69 Another example is the lethal-7 (let-7) gene, whose silencing is directly associated with a BCSC phenotype.Citation70 The epigenetic silencing of let-7e mediated by the histone H3K4me3 demethylase JARID1B, over-expressed in breast cancer, contributes to cell cycle progression and the proliferation of cancer cells.Citation71,Citation72 Over-expression of other epigenetic modifiers can also induce BCSC plasticity. For instance, high levels of the polycomb protein EZH2 have been found in high grade breast cancers, and it plays a fundamental role in the regulation of stem cell self-renewal and differentiation.Citation73,Citation74
The type of epigenetic alterations involved in the generation of BCSCs may depend on their normal cell-of-origin, and therefore different cancer subtypes could acquire clonal characteristics. This is demonstrated by the fact that transformation of genetically identical but phenotypically distinct breast epithelial cells can result in different cancer types.Citation15,Citation75
Can we target BCSC plasticity? The ultimate answer to the ultimate question
Tumor heterogeneity presents a major clinical problem. Important hurdles for successful treatment approaches include treating tumor subtypes with specific therapies and targeting elusive BCSCs. Breast cancer management involves a combination of surgery, chemotherapy, radiotherapy, and some targeted therapies such as hormonal therapy and the use of monoclonal antibodies.Citation2 Although chemotherapy and radiotherapy are able to debulk the tumor mass, the majority of patients with basal or HER2+ cancer subtypes will relapse due to a minimal residual disease (MRD). BCSCs are directly implicated in MRD because of their intrinsic and extrinsic characteristics that are ultimately responsible for tumor recurrence.Citation20 CSCs have an innate chemo- and radio-resistance due to the expression of drug transporters and detoxifying enzymes; altered DNA damage response mechanisms, such as resistance to apoptosis; enhanced DNA repair mechanisms; and quenching of reactive oxygen species.Citation20 Stochastic events and adjuvant chemotherapy treatments can act as external factors that can generate resistant BCSCs clones. Such foci of surviving cells are subject to selective pressure and may lead to the development of complex drug resistance mechanisms and increased aggressiveness of the resistant cells (it should be noted that drug resistance can involve endocrine therapies as well).Citation76 Hormonal therapy fails to target ER−BCSCs, but they also induce the conversion of cancer cells to BCSCs.Citation77 Since the adaptation and selection of different BCSC clones depend on cancer treatments, it is essential that future research efforts employ cancer patient stratification for more personalized treatment approaches.
Many studies are now focusing on targeting BCSC signaling pathways involved in stem cell self-renewal, such as Notch, Hedgehog, and Wnt; these have been extensively reviewed elsewhere.Citation78–Citation80 Other strategies have considered targeting EMT, which can be at the core of BCSC plasticity. Even if limited to the targeting of BCSCs with a basal phenotype, this strategy is of considerable interest;Citation81 however, targeting of EMT on its own may not be a sufficient approach, as a crosstalk between embryonic signaling pathways exists. For instance, the TGFβ pathway, a main inducer of EMT, is known to interact with Wnt, Notch, and Hedgehog.Citation78 There is also evidence that ER signaling influences EMT and the induction of BCSCs.Citation77
The tumor microenvironment should also be considered for the development of BCSC-targeted therapies, and integrin-related signaling components, BCSC-surface markers, and stroma-secreted cytokines could be potential targets.Citation82 The developmental plasticity of BCSCs as well as the reversible nature of the epigenetic alterations that regulate their function have led to the development of epigenetic therapies as new treatment options. Because epigenetic drugs can restore normal tissue homeostasis, they are a promising tool for differentiation therapyCitation83 The idea that BCSCs can be reset to their normal function via modification of their epigenetic landscape is a desired prospect. Tumorigenicity of breast cancer cells harboring genetic defects can be abolished by epigenetic reprogramming, leading to reactivation of silenced tumor suppressor genes.Citation84 Therefore, one can envision a therapeutic approach aimed at modulating dominant non-genetic defects to control stem cell function.
Epigenetic drugs that inhibit DNA methylation (DNMT inhibitors) and histone deacetylation (HDAC inhibitors) can restore the expression of silenced tumor suppressor genes, and these drugs are particularly effective for the treatment of leukemia. For instance, the DNA demethylating agent 5-aza-2′-deoxycydine (AZA) has been shown to inhibit self-renewal of leukemic CSCs.Citation85 In 2009, Stand Up to Cancer began testing epigenetic drugs on BCSCs and assessed their efficacy for the treatment of HER2+ and triple-negative breast cancer in clinical trials. Preclinical studies have shown that two demethylating agents, Decitabine and AZA at low and transient doses, induce the inhibition of BCSC growth as mammospheres due to a decrease in the CD44+/CD24−/ALDH1+ stem cell population across breast cancer cell lines. This treatment can also reduce the growth of patient-derived BCSC tumor xenografts due the underlying reactivation of tumor suppressor genes, and also due to the alteration of major cancer cell signaling pathways.Citation86 These exciting results suggest that epigenetic therapies can become a reality as they can directly target BCSCs both in vitro and in vivo.
Conclusion
Breast cancer heterogeneity profoundly impacts the clinical management of the disease. Because of the inter- and intraheterogeneity of breast tumors, classical and targeted therapies are not always successful in eradicating the disease, resulting in poor patient outcome. Cancer recurrence and metastasis are the main cause of poor patient survival, both of which are caused by expansion of MRD. Many studies have shown that tumor heterogeneity arises from BCSCs, whereas others have demonstrated that it is actually the result of clonal evolution. While BCSC and clonal evolution studies show extensive evidence in support of their arguments, caveats in both models still remain. A unified model based on stem cell plasticity can reconcile both views and account for clinical and molecular characteristics of different breast tumor subtypes. According to the stem cell plasticity model, tumor progression is highly dynamic, with cancer cells constantly exposed to internal and external survival pressures. In this context, different areas of the tumor are affected differently by the microenvironment. Therefore, cells may be selected according to clonal evolution, stochastic plasticity, or according to the BCSC differentiation hierarchy at different times and in different regions of the tumor ().
Careful assessments of tumor heterogeneity and its microenvironment are therefore needed to devise targeted therapeutic strategies, as generalized interventions can result in the selection of the most resistant cancer cell population and recurrence of a more aggressive form of the disease. Cancer cell plasticity presents a clinical challenge, and future research should focus on understanding how to control its intrinsic and extrinsic effectors; this is certainly not an easy quest and novel avenues should be considered. In this light, epigenetic therapies may offer new solutions. By reversing the epigenetic landscape of cancer cells, epigenetic drugs can reset cancer cell plasticity and cause cancer cells to revert to normalcy. This approach might be instrumental as a new approach in the management of the disease, insofar as cancer could be treated as a chronic condition.
Disclosure
The authors report no conflicts of interest in this work.
Acknowledgment
The authors acknowledge the University of Nottingham, the Royal Society of London, and Evocell Ltd for funding.
Figure 1 Models explaining the origins of tumor heterogeneity in breast cancer.
Abbreviation: BCSC, breast cancer stem cell.
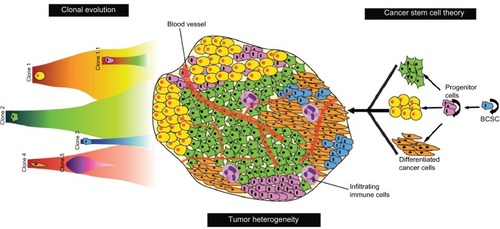
Figure 2 Proposed model of the human MaSC differentiation hierarchy with corresponding surface markers for stem/progenitor cell identification and isolation.
Abbreviation: MaSC, mammary stem cell.
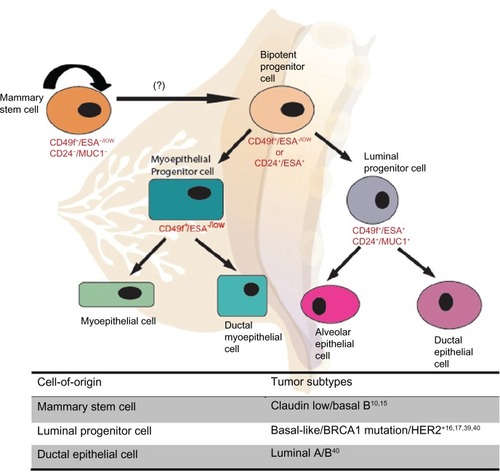
Figure 3 Unified theory of tumor heterogeneity.
Abbreviations: BCSC, breast cancer stem cells; ECM, extracellular matrix.
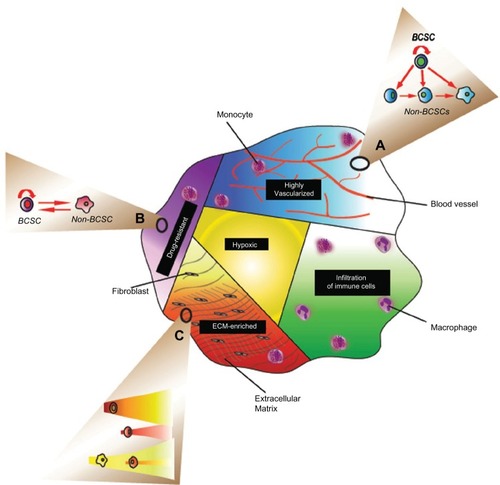
Table 1 Biological and clinical characteristics of breast cancer molecular subtypes
References
- JemalABrayFCenterMMFerlayJWardEFormanDGlobal cancer statisticsCA Cancer J Clin2011612699021296855
- AlmendroVFusterGHeterogeneity of breast cancer: etiology and clinical relevanceClin Transl Oncol2011131176777322082639
- TavassoliFAMillisRRBoeckerWLakhaniSRLobular NeoplasiaTavassoliFADevileePWorld Health Organization Classification of TumoursLyon, FranceIARC Press20036062
- Reis-FilhoJSPusztaiLGene expression profiling in breast cancer: classification, prognostication, and predictionLancet201137898051812182322098854
- HuangTHEstellerMChromatin remodeling in mammary gland differentiation and breast tumorigenesisCold Spring Harb Perspect Biol201029a00451520610549
- HanahanDWeinbergRAHallmarks of cancer: the next generationCell2011144564667421376230
- SorlieTPerouCMTibshiraniRGene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implicationsProc Natl Acad Sci U S A20019819108691087411553815
- SorlieTTibshiraniRParkerJRepeated observation of breast tumor subtypes in independent gene expression data setsProc Natl Acad Sci U S A2003100148418842312829800
- SotiriouCNeoSYMcShaneLMBreast cancer classification and prognosis based on gene expression profiles from a population-based studyProc Natl Acad Sci U S A200310018103931039812917485
- PerouCMSorlieTEisenMBMolecular portraits of human breast tumoursNature2000406679774775210963602
- CalzaSHallPAuerGIntrinsic molecular signature of breast cancer in a population-based cohort of 412 patientsBreast Cancer Res200684R3416846532
- PratAPerouCMDeconstructing the molecular portraits of breast cancerMol Oncol20115152321147047
- PodoFBuydensLMDeganiHFEMME ConsortiumTriple-negative breast cancer: present challenges and new perspectivesMol Oncol20104320922920537966
- CurtisCShahSPChinSFMETABRIC GroupThe genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroupsNature2012486740334635222522925
- KellerPJArendtLMSkibinskiADefining the cellular precursors to human breast cancerProc Natl Acad Sci U S A201210982772277721940501
- LimEVaillantFWuDAberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriersNat Med200915890791319648928
- MolyneuxGGeyerFCMagnayFABRCA1 basal-like breast cancers originate from luminal epithelial progenitors and not from basal stem cellsCell Stem Cell20107340341720804975
- AllredDCBrownPMedinaDThe origins of estrogen receptor alphapositive and estrogen receptor alpha-negative human breast cancerBreast Cancer Res20046624024515535853
- ShahMAllegrucciCStem cell plasticity in development and cancer: epigenetic origin of cancer stem cellsKunduTKEpigenetics: Development and DiseaseSpringerSubcellular Biochemistry201261
- VisvaderJELindemanGJCancer stem cells in solid tumours: accumulating evidence and unresolved questionsNat Rev Cancer200881075576818784658
- BadveSNakshatriHBreast-cancer stem cells-beyond semanticsLancet Oncol2012131e43e4822225725
- StinglJEirewPRicketsonIPurification and unique properties of mammary epithelial stem cellsNature2006439707999399716395311
- ShackletonMVaillantFSimpsonKJGeneration of a functional mammary gland from a single stem cellNature20064397072848816397499
- Van KeymeulenARochaASOussetMDistinct stem cells contribute to mammary gland development and maintenanceNature2011479737218919321983963
- EirewPStinglJRaoufAA method for quantifying normal human mammary epithelial stem cells with in vivo regenerative abilityNat Med200814121384138919029987
- RaoufASunYChatterjeeSBasakPThe biology of human breast epithelial progenitorsSemin Cell Dev Biol201223560661222609813
- ChenJLiYYuTSA restricted cell population propagates glioblastoma growth after chemotherapyNature2012488741252252622854781
- Al-HajjMWichaMSBenito-HernandezAMorrisonSJClarkeMFProspective identification of tumorigenic breast cancer cellsProc Natl Acad Sci U S A200310073983398812629218
- ShipitsinMCampbellLLArganiPMolecular definition of breast tumor heterogeneityCancer Cell3200711325927317349583
- PontiDCostaAZaffaroniNIsolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell propertiesCancer Res200565135506551115994920
- FillmoreCMKuperwasserCHuman breast cancer cell lines contain stem-like cells that self-renew, give rise to phenotypically diverse progeny and survive chemotherapyBreast Cancer Res2008102R2518366788
- GinestierCHurMHCharafe-JauffretEALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcomeCell Stem Cell20071555556718371393
- EirewPKannanNKnappDJAldehyde dehydrogenase activity is a biomarker of primitive normal human mammary luminal cellsStem Cells201230234434822131125
- DontuGAbdallahWMFoleyJMIn vitro propagation and transcriptional profiling of human mammary stem/progenitor cellsGenes Dev200317101253127012756227
- HarrisonHFarnieGBrennanKRClarkeRBBreast cancer stem cells: something out of notching?Cancer Res201070228973897621045140
- PeceSTosoniDConfalonieriSBiological and molecular heterogeneity of breast cancers correlates with their cancer stem cell contentCell20101401627320074520
- Charafe-JauffretEGinestierCBirnbaumDBreast cancer stem cells: tools and models to rely onBMC Cancer2009920219555472
- DriessensGBeckBCaauweASimonsBDBlanpainCDefining the mode of tumour growth by clonal analysisNature2012488741252753022854777
- ProiaTAKellerPJGuptaPBGenetic predisposition directs breast cancer phenotype by dictating progenitor cell fateCell Stem Cell20118214916321295272
- VisvaderJECells of origin in cancerNature2011469733031432221248838
- SchepersAGSnippertHJStangeDELineage tracing reveals Lgr5+ stem cell activity in mouse intestinal adenomasScience2012337609573073522855427
- GilbertsonRJGrahamTACancer: Resolving the stem-cell debateNature2012488741246246322919708
- KernSEShibataDThe fuzzy math of solid tumor stem cells: a perspectiveCancer Res200767198985898817908998
- Ronnov-JessenLPetersenOWBissellMJCellular changes involved in conversion of normal to malignant breast: importance of the stromal reactionPhysiol Rev1996761691258592733
- LindemanGJVisvaderJEInsights into the cell of origin in breast cancer and breast cancer stem cellsAsia Pac J Clin Oncol201062899720565420
- QuintanaEShackletonMSabelMSFullenDRJohnsonTMMorrisonSJEfficient tumour formation by single human melanoma cellsNature2008456722259359819052619
- StinglJDetection and analysis of mammary gland stem cellsJ Pathol2009217222924119009588
- MeyerMJFlemingJMAliMAPeseskyMWGinsburgEVonderhaarBKDynamic regulation of CD24 and the invasive, CD44posCD24neg phenotype in breast cancer cell linesBreast Cancer Res2009116R8219906290
- CicaleseABonizziGPasiCEThe tumor suppressor p53 regulates polarity of self-renewing divisions in mammary stem cellsCell200913861083109519766563
- NowellPCThe clonal evolution of tumor cell populationsScience197619442602328959840
- SpencerSLGaudetSAlbeckJGBurkeJMSorgerPKNon-genetic origins of cell-to-cell variability in TRAIL-induced apoptosisNature2009459724542843219363473
- MarusykAAlmendroVPolyakKIntra-tumour heterogeneity: a looking glass for cancer?Nat Rev Cancer201212532333422513401
- MarusykAPolyakKTumor heterogeneity: causes and consequencesBiochim Biophys Acta20101805110511719931353
- NavinNKendallJTrogeJTumour evolution inferred by single-cell sequencingNature20114727341909421399628
- ShahSPRothAGoyaRThe clonal and mutational evolution spectrum of primary triple-negative breast cancersNature2012486740339539922495314
- ShahMAllegrucciCStem cell plasticity in development and cancer: epigenetic origin of cancer stem cellsKunduTKEpigenetics: Development and Disease61New YorkSpringer2012
- AllegrucciCWuYZThurstonARestriction landmark genome scanning identifies culture-induced DNA methylation instability in the human embryonic stem cell epigenomeHum Mol Genet200716101253126817409196
- ShenYChowJWangZFanGAbnormal CpG island methylation occurs during in vitro differentiation of human embryonic stem cellsHum Mol Genet200615172623263516870691
- WidschwendterMFieglHEgleDEpigenetic stem cell signature in cancerNat Genet200739215715817200673
- OhmJEMcGarveyKMYuXA stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencingNat Genet200739223724217211412
- SchlesingerYStraussmanRKeshetIPolycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancerNat Genet200739223223617200670
- GuptaPBFillmoreCMJiangGStochastic state transitions give rise to phenotypic equilibrium in populations of cancer cellsCell2011146463364421854987
- ChafferCLBrueckmannIScheelCNormal and neoplastic nonstem cells can spontaneously convert to a stem-like stateProc Natl Acad Sci U S A2011108197950795521498687
- Ben-PorathIThomsonMWCareyVJAn embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumorsNat Genet200840549950718443585
- ManiSAGuoWLiaoMJThe epithelial-mesenchymal transition generates cells with properties of stem cellsCell2008133470471518485877
- PolyakKWeinbergRATransitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traitsNat Rev Cancer20099426527319262571
- Bloushtain-QimronNYaoJSnyderELCell type-specific DNA methylation patterns in the human breastProc Natl Acad Sci U S A200810537140761408118780791
- HermekingHMicroRNAs in the p53 network: micromanagement of tumour suppressionNat Rev Cancer201212961362622898542
- ShimonoYZabalaMChoRWDownregulation of miRNA-200c links breast cancer stem cells with normal stem cellsCell2009138359260319665978
- ZimmermanALWuSMicroRNAs, cancer and cancer stem cellsCancer Lett20113001101920965651
- MitraDDasPMHuynhFCJonesFEJumonji/ARID1 B (JARID1B) protein promotes breast tumor cell cycle progression through epigenetic repression of microRNA let-7eJ Biol Chem201128647405314053521969366
- BarrettASantangeloSTanKBreast cancer associated transcriptional repressor PLU-1/JARID1B interacts directly with histone deacetylasesInt J Cancer2007121226527517373667
- BrackenAPDietrichNPasiniDHansenKHHelinKGenome-wide mapping of Polycomb target genes unravels their roles in cell fate transitionsGenes Dev20062091123113616618801
- HolmKGrabauDLovgrenKGlobal H3K27 trimethylation and EZH2 abundance in breast tumor subtypesMol Oncol20126549450622766277
- InceTARichardsonALBellGWTransformation of different human breast epithelial cell types leads to distinct tumor phenotypesCancer Cell200712216017017692807
- MeadsMBGatenbyRADaltonWSEnvironment-mediated drug resistance: a major contributor to minimal residual diseaseNat Rev Cancer20099966567419693095
- SmalleyMPiggottLClarksonRBreast cancer stem cells: Obstacles to therapyCancer Lett4 302012 Epub ahead of print
- TakebeNHarrisPJWarrenRQIvySPTargeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathwaysNat Rev Clin Oncol2011829710621151206
- ZhouBBZhangHDamelinMGelesKGGrindleyJCDirksPBTumour-initiating cells: challenges and opportunities for anticancer drug discoveryNat Rev Drug Discov200981080682319794444
- AlisonMRLimSMNicholsonLJCancer stem cells: problems for therapy?J Pathol2011223214716121125672
- KimJVilladsenRSorlieTTumor initiating but differentiated luminal-like breast cancer cells are highly invasive in the absence of basal-like activityProc Natl Acad Sci U S A2012109166124612922454501
- AblettMPSinghJKClarkeRBStem cells in breast tumours: Are they ready for the clinic?Eur J Cancer201248142104211622542086
- BerdascoMEstellerMAberrant epigenetic landscape in cancer: how cellular identity goes awryDev Cell201019569871121074720
- AllegrucciCRushtonMDDixonJEEpigenetic reprogramming of breast cancer cells with oocyte extractsMol Cancer2011101721232089
- HuZNegrottoSGuXDecitabine maintains hematopoietic precursor self-renewal by preventing repression of stem cell genes by a differentiation-inducing stimulusMol Cancer Ther2010961536154320501800
- TsaiHCLiHVan NesteLTransient low doses of DNA-demethylating agents exert durable antitumor effects on hematological and epithelial tumor cellsCancer Cell201221343044622439938