Abstract
In our previous study, we identified that a protein target, peroxiredoxin II (PrxII), is overexpressed in radioresistant MCF+FIR3 breast-cancer cells and found that its expression and function is associated with breast-cancer radiation sensitivity or resistance. Small interference RNA (siRNA) targeting PrxII gene expression was able to sensitize MCF+FIR3 radioresistant breast-cancer cells to ionizing radiation. The major focus of this work was to investigate how the radiation response of MCF+FIR3 radioresistant cells was affected by the siRNA that inhibits PrxII gene expression. Our results, presented here, show that silencing PrxII gene expression increased cellular toxicity by altering cellular thiol status, inhibiting Ca2+ efflux from the cells, and perturbing the intracellular Ca2+ homeostasis. By combining radiotherapy and siRNA technology, we hope to develop new therapeutic strategies that may have potential to enhance the efficacy of chemotherapeutic agents due to this technology’s property of targeting to specific cancer-related genes.
Keywords:
Introduction
Metastatic and recurrent breast cancer are distinct from early stage breast cancer.Citation1 As a result, the recurrent form of the disease is resistant to existing targeted treatments, and the mortality rate is thus significantly higher, with as many as 40% relapsing with metastatic disease.Citation1–Citation3 The biological mechanisms underlying intrinsic and acquired resistance to chemotherapy and radiation therapy are therefore of considerable clinical significance. Studies have shown that breast-cancer cells that are able to propagate as mammospheres and possess cancer stem cell (CSC) properties are more radioresistantCitation4 and the population of breast-cancer stem cells (BCSCs) is increased after chemotherapy.Citation5 Therefore, the design of therapies targeting specific genes and proteins actively engaged in the physiopathology of radioresistant breast cancer is greatly demanded. Two-dimensional electrophoresis combined with mass spectrometry has been widely used to study protein expression profiles and to identify possible therapeutic targets. Using two-dimensional proteomic technology, we have identified a peroxiredoxin (Prx) II that plays an important role in the development of radioresistant breast-cancer cells that are enriched with a subpopulation of BCSCs.Citation6 “PrxII” is a member of a ubiquitous family of antioxidant enzymes that normally respond to environmental oxidative stress or reactive oxygen species (ROS).Citation7–Citation9 Unchecked ROS can lead to oxidative damage in the cell, in turn leading to lipid peroxidation, protein modification, DNA modification, and strand breaks. Prxs play a central role in the cellular defense against oxidative damage through their peroxidase activity, whereby hydrogen peroxide, peroxynitrite, and a wide range of organic hydroperoxides are reduced and detoxified.Citation10–Citation12
The mechanism of detoxification by PrxII is the reduction of peroxides via cysteine residue in the active site. As a result of ionizing radiation (IR) or other environmental oxidative stress, ROS lead to the rapid inactivation of PrxII by the modification of the catalytic cysteine (R-SH) thiol to an inactive cysteine disulfide (R-S-S-R). This modification of the cysteine thiol to an inactive cysteine disulfide is completely reversible by the subsequent reduction of the inactive cysteine disulfide to regenerate the active PrxII cysteine.Citation13 Evidence points to a pivotal role for PrxII in the cellular defense system. PrxII-overexpressing cells are more resistant to the environmental oxidative damage caused by inorganic hydrogen peroxide, organic t-butyl hydrogen peroxide, the heavy metal mercury, and anticancer drugs like cisplatin.Citation14–Citation17 Thus, PrxII is a promising candidate for gene-targeted therapy. In our previous study, small interference RNA (siRNA) was transfected into the radioresistant breast-cancer cells and was found to silence the gene expression of PrxII, resulting in enhanced radiosensitivity.
In the study reported here, we were interested in elucidating the mechanism by which inhibition by siRNA-PrxII of PrxII gene expression affects radiation-induced cell death in MCF+FIR3 radioresistant breast-cancer cells. Our hypothesis was that therapeutic IR combined with siRNA would enhance radiation-induced cell death by combining the gene-targeting property of siRNA and the cytotoxicity of radiation therapy. Our results showed that loss of protein thiol (PSH) and glutathione (GSH) (as a consequence of PrxII gene expression being silenced by PrxII-siRNA) is associated with increases in cellular toxicity, inhibition of Ca2+ efflux, and perturbation of Ca2+ homeostasis and results in enhanced radiosensitivity in MCF+FIR3 resistant cells. Thus, by combining radiotherapy with siRNA technology, we hope in future to be able to target cancer cells with specific genes involved in resistance. The results of these studies will be useful to help design advanced therapeutic strategies.
Materials and methods
Development of a radioresistant cell line
MCF-7 human breast-cancer cells were purchased from the American Type Culture Collection (Manassas, VA, USA). The MCF+FIR30 cell line was developed as described in a previous publication.Citation6 A radiation-resistant clone (MCF+FIR3) and a radiation-sensitive clone (MCF+FIS4) isolated from the MCF+FIR30 clonal populations were used for this study.
Gene targeting by siRNA
siRNA capable of targeting messenger RNAs (mRNAs) encoding PrxII was transfected using the same protocol as described before.Citation6 The sequences of five PrxII-siRNA sense strands were:
The sequence of the control siRNA was 5′-UCACUCCUAUAUCUUCUGUTT-3′. This was used as a control for the PrxII gene-targeting experiments.
Immunoblot analysis
Immunoblot analysis was carried out using the standard protocol as described previously.Citation18 PrxI, -II, and -III antibodies were used for these experiments. The immunoblot assays ran under the nonreducing experiments were performed in the absence of dithiothreitol (DTT) reducing reagent. Total protein concentration from the cell lysate was determined by Pierce™ BCA assay kit (Thermo Fisher Scientific, Waltham, MA, USA). Scanned images were quantified by Kodak 1D Image Analysis software, (Kodak, Rochester, NY, USA), standardized by β-actin. Microsoft® Excel 2007 software (Microsoft Corporation, Redmond, WA, USA) was used for data analysis.
3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide (MTT) toxicity assay
The MTT cytotoxicity method was used for cell viability measurements in multiwall plate format. Each experiment included a blank containing all of the reagents in a well without cells. MTT was dissolved in Dulbecco’s modified Eagle’s medium (DMEM) at 5 mg/mL and filtered through 0.22 μm filters before 1 × 107 viable cells/mL was suspended in culture medium. Following this, 100 μL of the solution was dispensed per well in 96-well flat-bottomed tissue-culture plates. Cells were allowed to grow with and without PrxII and control siRNA to determine the difference between cell proliferations. The 96-well plates were incubated at 37°C in an incubator for 48 hours. After this, the medium in each well was removed and 100 μL fresh medium added. MTT solution (10 μL) was then added to each well and the plates incubated for another 4 hours at 37°C. During this period, formazan crystals formed at the bottom of the wells. The solution in each well was aspirated and 100 μL dimethyl sulfoxide (DMSO) was added to each well and mixed thoroughly to dissolve the crystals. Absorbance was read at 570 nm. The experiments were repeated at least three times. Cell proliferation was calculated as proliferation percentage:
in which “ABS” represents absorbance.
Calcium assay
Determination of calcium content was accomplished by fluorometric determination. Cell pellets were suspended in a Ca2+ and Mg2+-free Hanks’ solution. Intracellular Ca2+ concentration was determined by fluorescent assay using a PerkinElmer fMax microplate reader (Molecular Devices, Sunnyvale, CA, USA) and a Twinkle LB 970 Microplate Fluorometer reader (Berthold Technologies GmbH & Co., Bad Wildbad, Germany). The method is based on the formation of a fluorescent chelate between Ca2+ and calcein fluorophore. The calcein solution was made by dissolving 0.01 mM calcein in a 0.8 M potassium hydroxide solution. The experiments were repeated at least three times.
PSH and GSH assay
The cell pellets were sonicated in 1 mL of cold buffer (50 mM phosphate, pH 6–7, containing 1 mM ethylenediaminetetraacetic acid [EDTA]). They were then centrifuged at 10,000 g for 15 minutes at 4°C. The supernatant was removed, stored on ice, and treated with 7% sulfosalicylic acid containing 0.1 mM EDTA. The mixture was centrifuged at maximum speed for 10 minutes at 4°C. Afterwards, the supernatant was collected for GSH assay. The precipitated pellets were washed three times with 7% sulfosalicylic acid containing 0.1 mM EDTA buffer. The denatured protein was dissolved in 0.1 M pH 7.5 phosphate buffer containing 0.1 mM EDTA to assay for PSH concentration. Total levels of PSH and GSH were measured using a Glutathione Assay Kit from Cayman Chemical (Ann Arbor, MI, USA), which utilizes a carefully optimized enzymatic recycling method to quantify the sulfhydryl group level. The sulfhydryl group of PSH and GSH reacts with 5,5′-dithio-bis-2-nitrobenzoic acid (DTNB; Ellman’s reagent) and produces a yellow-pigmented 5-thio-2-nitrobenzoic acid (TNB). Measurement of the absorbance of TNB at 405 nm provides an accurate estimation of PSH and GSH in the sample. The experiments were repeated at least three times. The experiment was performed in quadruple replicates each time.
Phosphate assay
Quantitation of inorganic phosphate (Pi) in solution was performed using a Phosphate Fluorometric Assay Kit from BioVision, Inc. (Milpitas, CA, USA). In this assay, Pi reacted with maltose to produce glucose in the presence of a special enzyme. The glucose is specifically oxidized to generate a product that reacts with the OxiRed™ fluorophore to generate fluorescence with the excitation wavelength at 535 nm and the emission wavelength at 587 nm. The fluorescent assay was performed using the Twinkle LB 970 Microplate Fluorometer reader. The experiments were repeated at least two times.
Results
Prx PSH status in radioresistant breast-cancer cells
PrxII belongs to a family of detoxification enzymes that use cysteine as an active site to reduce peroxides. PrxI, -II, and -III isozymes are typical 2-Cysteine (2Cys) Prxs and contain two conserved cysteine residues, the peroxidatic cysteine (generally near residue 50), and the resolving cysteine (near residue 170).Citation19 During the catalysis, the peroxidatic cysteine, a redox-sensitive reactive site, becomes oxidized, forming disulfide in the presence of ROS generated by an oxidative environment. GSH has been known to provide the cells with a reducing environment in addition to maintaining the proteins in a reduced state.Citation20 The oxidized PrxII is then reduced in the reducing environment to regenerate the active PrxII. This may be an important redox regulatory mechanism through the alteration of cellular thiol. PrxII has been shown to be overexpressed in radioresistant breast-cancer cells. The thiol status of PrxII might be related to the cellular oxidative environment and cellular resistance level to the environmental oxidants. Here, we performed immunoblot analyses to analyze the Prx thiol status in MCF+FIR3 radioresistant breast-cancer cells and MCF+FIS4 radiosensitive cells using PrxI, -II, and -III antibodies in the presence and absence of 0.1 mM DTT reducing agent. The results are shown in .
Figure 1 Immunoblot analysis of peroxiredoxin (Prx) I, II, and III in MCF+FIS3 radiosensitive breast-cancer cells and MCF+FIR3 radioresistant breast-cancer cells in the presence and absence of dithiothreitol (DTT) reducing agent. Lane 1 is the result for MCF+FIS4 radiosensitive cells. Lane 2 is the result for MCF+FIR3 radioresistant cells. (A–C) Immunoblot results for PrxI, -II, and -III in the absence of DTT reducing agent; (D–G) immunoblot results for PrxI, -II, and -III in the presence of DTT reducing agent; (H) control experiment for the immunoblot analyses using the β-actin antibody.
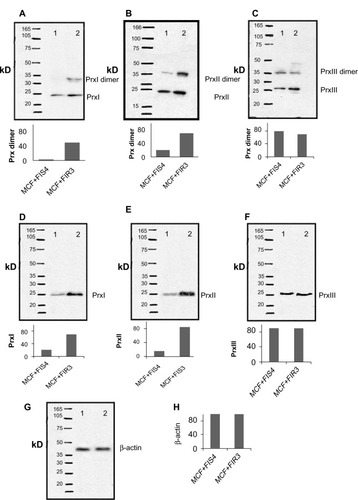
An increased amount of PrxI and PrxII dimers was observed in radioresistant MCF+FIR3 breast-cancer cells when compared with MCF+FIS4 radiosensitive cells ( and ). The dimers were formed through disulfide bonds between the peroxidatic cysteine in the first subunit and the resolving cysteine located at the C-terminus of another subunit in the nonreducing environment.Citation21 Unlike PrxI and -II, the level of PrxIII dimer was seen to be higher in the MCF+FIS4 radiosensitive cells (). It is likely that the location differentiates the response of PrxI, -II, and -III to IR. Both PrxI and PrxII are cytosolic proteins. PrxI is also found in the nucleus and PrxII is also associated with the membrane. PrxIII is exclusively located in the mitochondria. The higher dimerization level of PrxIII in MCF+FIS3 sensitive cells could possibly result from baseline exposure to the oxidative environment found in the mitochondria, even in the absence of radiation. The increased levels of PrxI and -II dimers in MCF+FIR3 radioresistant cells were mainly due to the prolonged exposure of MCF-7 cells to IR.
The immunoblot analyses in the reducing condition were performed alongside the just-mentioned experiments. – display the immunoblot results of PrxI, -II, and -III in MCF+FIS4 radiosensitive and MCF+FIR3 radioresistant cells. The inter-subunit disulfide bonds were reduced and PrxI, -II, and -III monomers generated. The control experiment for the immunoblot is shown in . The formation of intra- and inter-molecular disulfides between protein cysteines is believed to be associated with the stabilization of extracellular proteins, protection of proteins against irreversible oxidation of critical cysteine residues, and regulation of enzyme functions and transcription.Citation22 The results from this experiment suggest that the formation of Prx dimers in MCF+FIR3 radioresistant breast-cancer cells was an adaptive response of the MCF-7 breast-cancer cells to the oxidative environment created by chronic irradiation.
Cellular toxicity of MCF+FIR3 radioresistant cells after inhibition of PrxII expression
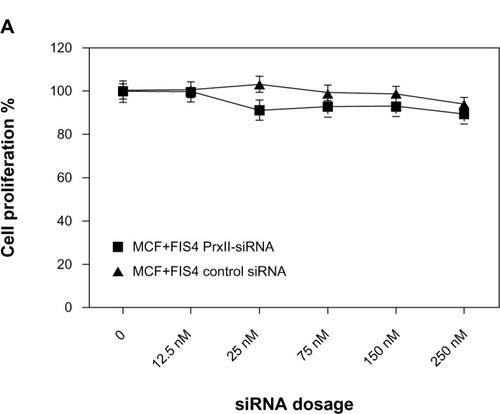
In the present study, the toxicological implication of PrxII-siRNA gene inhibition in MCF+FIR3 resistant and MCF+FIS4 sensitive cells was investigated. We were interested in studying how radioresistant and radiosensitive breast-cancer cells respond to IR when PrxII expression is knocked down. The siRNA was used to modulate the PrxII expression levels in the MCF+FIR3 and MCF+FIS4 cells in this experiment. Cytotoxicity assays of MCF+FIR3 radioresistant cells and MCF+FIS4 radiosensitive cells treated by control or PrxII-siRNA in the presence and absence of IR were examined. The results are shown in . We found that cell proliferation was not significantly affected by the treatment of PrxII-siRNA in MCF+FIS4 radiosensitive cells ( and ). This indicated that alteration of PrxII levels in MCF+FIS4 cells did not have any significant impact on cell proliferation or cell death. and display the results of cell proliferation for MCF+FIS4 radiosensitive cells treated with siRNA combined with IR. Cell proliferation in MCF+FIS4 cells pretreated with PrxII-siRNA or control siRNA was significantly reduced after IR. The reduced cell proliferation was mainly due to the radiation sensitivity of MCF+FIS4 cells (data not shown). PrxII-siRNA did not appear to play a significant role in the decrease of cell proliferation after IR, although the resistance relative to the sensitive cells was correlated.
Figure 2 Cell proliferation of MCF+FIS4 radiosensitive and MCF+FIR3 radioresistant breast-cancer cells in response to radiation and small interference RNA (siRNA) treatment. (A) Cell proliferation of MCF+FIS4 radiosensitive cells following the transfection of siRNA at concentrations 0.0, 12.5, 25.0, 75.0, 150.0, and 250.0 nM. (▲) Result for the cells treated with control siRNA. (■) Result for the cells treated with peroxiredoxin (Prx) II-siRNA. (B) Immunoblot results for β-actin and PrxII from cells transfected with control siRNA and cells transfected with PrxII-siRNA. (C) Cell proliferation results for MCF+FIS4 radiosensitive cells with combined treatment of 25 nM siRNA for 72 hours and ionizing radiation (IR). (♦) Result for the cells treated with combined PrxII-siRNA and IR. (▲) Result for cells treated with combined control siRNA and IR. (■) Result for the MCF+FIR3-resistant cells used for comparison analysis. (D) Immunoblot analysis undertaken to monitor PrxII expression levels after control siRNA and PrxII-siRNA transfection. (E) Cell proliferation for MCF+FIR3 radiosensitive cells following the transfection of siRNA at concentrations 0.0, 12.5, 25.0, 75.0, 150.0, and 250.0 nM. (▲) Result for the cells treated with control siRNA. (■) Result for the cells treated with PrxII-siRNA. (F) PrxII expression levels were monitored by immunoblot analysis. (G) Cell proliferation results for MCF+FIR3 radioresistant cells treated with combined siRNA (25 nM) for 72 hours and IR. (•) Result for the cells treated with combined PrxII-siRNA and IR. (■) Result for cells treated with control siRNA and IR. (♦) Cell proliferation results for untreated MCF+FIR3 radioresistant cells. (H) PrxII expression levels were monitored by immunoblot analyses after control siRNA and PrxII-siRNA transfection.
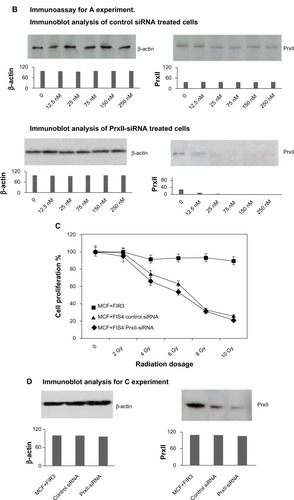
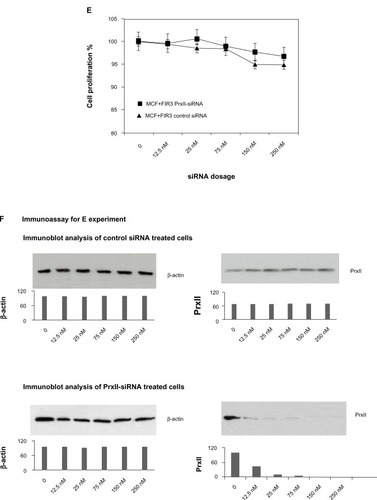
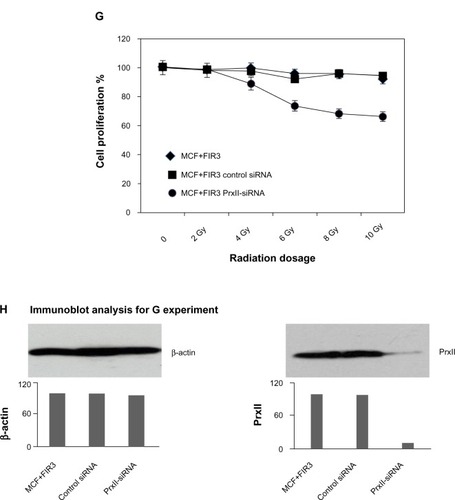
displays the cell proliferation results in MCF+FIR3 radioresistant breast-cancer cells treated with control and PrxII-siRNA. PrxII expression levels were monitored by immunoblot analyses (). There was no significant change in cell proliferation for PrxII-siRNA treatment versus control siRNA treatment in MCF+FIR3 resistant cells. However, combined treatment with PrxII-siRNA and IR did enhance the cellular toxicity induced by radiation in comparison to the control experiments in MCF+FIR3 cells, as shown in . The immunoblot analyses ensured the PrxII expression was suppressed by PrxII-siRNA (). The results indicated that inhibition of PrxII gene expression by siRNA greatly reduced cell viability in response to IR. The combination of PrxII-siRNA and radiation treatment, which increased cellular toxicity induced by radiation, resulted in the increased radiosensitivity of MCF+FIR3 breast-cancer cells.
Cellular toxicity and cellular thiol status in the radioresistant breast-cancer cells after silencing PrxII gene expression
GSH is a major player in maintaining the cellular reducing environment. A PrxII dimer with a disulfide linkage can be reduced in the reducing environment with the presence of GSH. GSH and PSH have been shown to be associated with many redox-inducing reagents and chemotherapeutic drugs. We were interested in discovering whether the PrxII-siRNA that inhibits PrxII gene expression would affect the GSH and PSH levels and to see if these changes were directly reflected in intracellular thiol alterations. Since pretreatment with PrxII-siRNA did not show significant effects on cell proliferation in MCF+FIS4 radiosensitive cells in response to IR (), the focus of this study was the radiation response of MCF+FIR3 radioresistant cells under the condition of inhibited PrxII expression. We hypothesized that there may be a relationship between PrxII level and cellular PSH and GSH levels in MCF+FIR3 radioresistant cells.
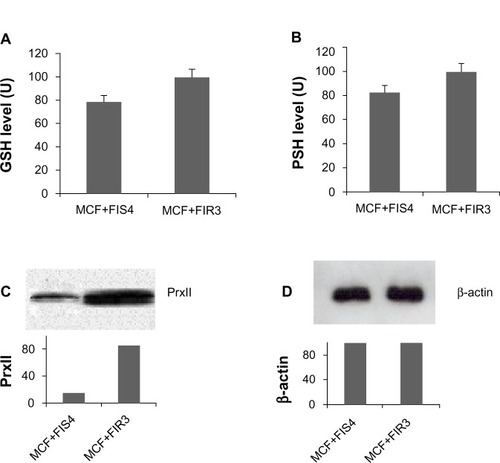
siRNA technology has become a powerful research tool to target molecules that conventional therapeutics cannot and is being actively pursued for its therapeutic potential.Citation23–Citation25 In this experiment, PrxII-siRNA was used to silence PrxII gene expression in comparison to the control siRNA that did not interfere with PrxII expression. A TNB assay was used to measure the cellular PSH and GSH levels. The disulfide was reduced by GSH reductase to recycle the PSH and GSH and produce more TNB. The rate of TNB production was directly proportional to this recycling reaction, which in turn was directly proportional to the concentration of PSH and GSH in the sample. shows the relative GSH and PSH levels in MCF+FIR3 radioresistant and MCF+FIS4 radiosensitive breast-cancer cells. The data were standard-ized by the total protein concentration of the cell extract. Relative GSH levels in MCF+FIR3 resistant and MCF+FIS4 sensitive cells are shown in , while PSH levels are shown in . Both GSH and PSH levels are higher in MCF+FIR3 resistant cells than in MCF+FIS4 radiosensitive cells. This result correlates well with the increased PrxII level in the MCF+FIR3 radioresistant cells in comparison to the MCF+FIS4 sensitive cells. PrxII expression levels in radioresistant and radiosensitive cells are shown in and . The lower GSH, PSH, and PrxII levels in MCF+FIS4 cells could be one of the factors that renders the cells more sensitive to radiation.
Figure 3 Glutathione (GSH) and protein thiol (PSH) levels in radioresistant breast-cancer cells and radiosensitive breast-cancer cells. (A) GSH levels in MCF+FIS4 sensitive cells and MCF+FIR3 resistant cells. (B) PSH levels in MCF+FIS4 sensitive cells and MCF+FIR3 resistant cells. (C) Immunoblot analysis of peroxiredoxin (Prx) II in MCF+FIS4 and MCF+FIR3 cells. (D) Immunoblot analysis of β-actin in MCF+FIS4 and MCF+FIR3 cells.
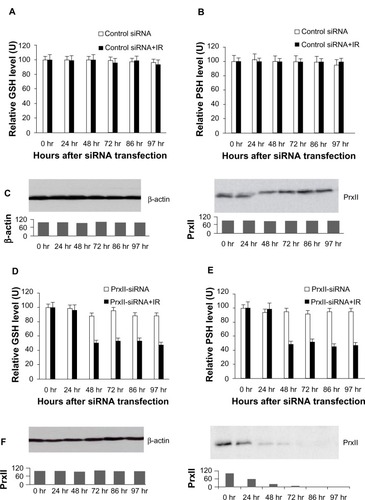
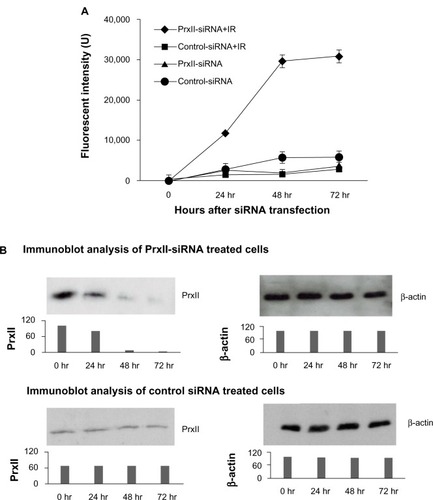
and display the results from the experiments in which the GSH and PSH levels in MCF+FIR3 radioresis-tant cells were measured after the treatment of control siRNA in comparison to combined control siRNA and IR treatment. The results showed that when PrxII was overexpressed in MCF+FIR3 resistant cells, the GSH and PSH levels were not affected significantly by IR treatment (). GSH and PSH levels in MCF+FIR3 radioresistant cells pretreated with PrxII-siRNA that inhibited PrxII gene expression are shown in and . In contrast to the result of the control siRNA experiment, GSH and PSH levels were significantly reduced by IR when PrxII gene expression was knocked down (). It appears that there is a connection between cellular radioresistance levels and cellular thiol status. The results indicate that the increased cellular toxicity – induced by IR in MCF+FIR3 radioresistant cells pretreated with PrxII-siRNA – that inhibited PrxII expression occurs through lowering cellular GSH and PSH levels.
Figure 4 Glutathione (GSH) and protein thiol (PSH) levels in MCF+FIR3 radioresistant cells pretreated with small interference RNA (siRNA), with and without subsequent 5 Gy ionizing radiation (IR). (A) GSH levels with control siRNA pretreatment, with and without subsequent 5 Gy IR. (□) Result for cells treated with control siRNA. (■) Result for cells treated with combined control siRNA and IR. (B) PSH levels pretreated with control siRNA, with and without subsequent 5 Gy IR. (□) Result for the cells treated with peroxiredoxin (Prx) II-siRNA. (■) Result for cells treated with combined PrxII-siRNA and IR. (C) Results of immunoblot analysis undertaken to monitor inhibition of PrxII expression by PrxII-siRNA. (D) GSH levels with PrxII-siRNA pretreatment, with and without subsequent 5 Gy IR. (□) Result for cells treated with PrxII-siRNA. (■) Result for cells treated with combined PrxII-siRNA and IR. (E) PSH levels with PrxII-siRNA pretreatment, with and without subsequent 5 Gy IR. (□) Result for cells treated with PrxII-siRNA. (■) Result for cells treated with combined PrxII-siRNA and IR. (F) Results of immunoblot analysis undertaken to monitor inhibition of PrxII expression by PrxII-siRNA.
Impairment of the plasma membrane Ca2+ pump in PrxII-siRNA-treated radioresistant breast-cancer cells as a consequence of perturbation of the cellular thiol homeostasis by IR
We were interested in investigating whether the loss of cellular thiol induced by IR as a result of PrxII-siRNA gene inhibition impairs the activity of the plasma membrane Ca2+ pump. The Ca2+ level was measured with sequential addition of carbonyl cyanide m-chlorophenyl hydrazone (CCCP), digitonin, and A23187 compounds. “CCCP” is a protonophore that dissipates the mitochondrial membrane potential, releases sequestered mitochondrial calcium stores, and irreversibly prevents mitochondrial calcium uptake. Digitonin makes the plasma membrane permeable without affecting the mitochondrial and endoplasmic reticular membrane, while the A23187 Ca2+ ionophore makes all cellular compartments permeable.
The rationale for this experiment was that cells were exposed to CCCP, which released Ca2+ from mitochondria to cytosol. The subsequent addition of digitonin would release the Ca2+ from cytosol to the extracellular space, while keeping the mitochondrial and endoplasmic reticular membranes intact. Adding A23187 would release the remaining intracellular Ca2+, thus allowing the measurement of the total cellular Ca2+ released.Citation26 As shown in , the total Ca2+ released from the cytosol by the sequential addition of CCCP, digitonin, and A23187 was not significantly affected in MCF+FIR3 radioresistant cells transfected with control siRNA after IR when PrxII gene expression was not inhibited (). This result shows that the released Ca2+ from the cytosol could be extruded into the extracellular space by membrane Ca2+-adenylpyrophosphatase (ATP) when PrxII was overexpressed in MCF+FIR3 resistant cells, suggesting that the plasma membrane pump Ca2+-ATPase was functional so could extrude the Ca2+ from cytosol to the extracellular space after IR.
Figure 5 Effect of calcium released from MCF+FIR3 radioresistant cells transfected with control small interference RNA (siRNA) and peroxiredoxin (Prx) II-siRNA in response to ionizing radiation (IR). (A) Calcium released in response to IR for MCR+FIR3 resistant cells transfected with 25 nM control siRNA. (■) Cells transfected with control siRNA in response to IR with sequential addition of the carbonyl cyanide m-chlorophenyl hydrazone (CCCP), A23187 (A), and digitonin (D). (◩) Cells transfected with control siRNA in response to IR with the sequential addition of CCCP and digitonin. (⊟) Cells transfected with control siRNA in response to IR with the addition of CCCP and A23187. (□) Cells transfected with control siRNA in response to IR with the addition of CCCP. (B) Results of immunoblot analysis undertaken to monitor PrxII expression by PrxII-siRNA. (C) Calcium released in response to IR for MCR+FIR3 resistant cells transfected with 25 nM PrxII-siRNA. (■) Cells transfected with PrxII-siRNA in response to IR with sequential addition of the CCCP, A23187, and digitonin. (◩) Cells transfected with PrxII-siRNA in response to IR with the sequential addition of CCCP and digitonin. (⊟) Cells transfected with PrxII-siRNA in response to IR with the addition of CCCP and A23187. (□) Cells transfected with PrxII-siRNA in response to IR with the addition of CCCP. (D) Results of immunoblot analysis undertaken to monitor inhibition of PrxII expression by PrxII-siRNA.
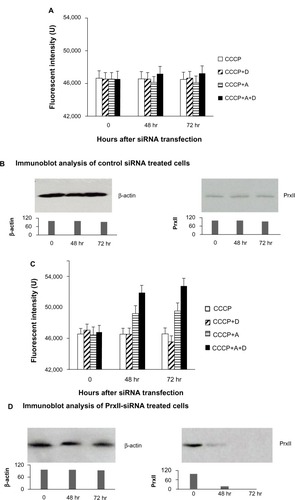
When PrxII gene expression was inhibited by PrxII-siRNA transfection, a significant increase in Ca2+ level was observed after IR in MCF+FIR3 radioresistant cells with the sequential addition of CCCP and A23187 ( and ). The increase in Ca2+ level was also seen in cells treated with A23187, CCCP, and digitonin (). The amount of Ca2+ mobilized by CCCP in MCF7+FIR3 radioresistant breast-cancer cells transfected by PrxII-siRNA, not excreted from the cells following IR, was markedly increased compared with in the control cells treated with the control siRNA, indicating that the plasma membrane Ca2+-extruding mechanism was inhibited. This result suggests that IR had a significant impact on the Ca2+ membrane extrusion mechanism after the silencing PrxII gene expression by PrxII-siRNA in MCF+FIR3 resistant cells.
Since plasma membrane Ca2+-ATPase is known to play a critical role in regulating cytosolic free Ca2+ concentration, it was of interest to investigate whether this impairment of Ca2+ efflux by PrxII-siRNA following IR was associated with an enhanced cytosolic free Ca2+ level. To monitor cytosolic Ca2+ level in MCF7+FIR3 radioresistant cells transfected with PrxII-siRNA following IR, the level of Pi was monitored. The Pi level in normal blood is very similar to that of calcium. The use of this activity as an index of cytosolic Ca2+ level fluctuations has been previously validated.Citation27 The addition and removal of phosphate from proteins in all cells is a pivotal strategy in the regulation of metabolic processes. As shown in and , an increase in the concentration of Pi was associated with IR treatment in MCF7+FIR3 radioresistant breast-cancer cells when PrxII expression was suppressed. The phosphate level in the radioresistant cells treated with control siRNA did not show any significant change; neither was there any significant change in phosphate level in the radioresistant cells treated with combined control siRNA and IR. Since PrxII was found to be associated with the cell membrane, the result may suggest that PrxII plays a role in protecting the cell membrane from IR-induced oxidative damage.
Figure 6 Intracellular phosphate levels in MCF+FIR3 radioresistant breast-cancer cells pretreated with 25 nM of small interference RNA (siRNA) in the presence and absence of ionizing radiation (IR). (A) Fluorescent intensity of intracellular phosphate levels measured at 535 nm and the emission wavelength at 587 nm. (♦) Cells treated with peroxiredoxin (Prx) II-siRNA and IR. (■) Cells treated with control siRNA and IR. (▲) Cells treated with PrxII-siRNA. (•) Cells treated with control siRNA. (B) Results of immunoblot analysis undertaken to monitor inhibition of PrxII expression by PrxII-siRNA.
Discussion
Advanced and recurrent tumors are usually resistant to chemotherapy and radiation therapy, leading to a poor survival rate. It has been reported that while traditional chemotherapy can kill the majority of cancer cells, it fails to target CSCs.Citation28 Moreover, initial treatment can increase the proportion of radio- and drug-resistant CSCs, resulting in disease recurrence.Citation29 Radiotherapy is still one of the most commonly used chemotherapeutic methods for cancer recurrence treatment. The problem with this method is its lack of specificity in targeting cancer cells. Here, we augmented therapeutic IR with a targeting property to recognize certain genes inside cancer cells that are activated in adaptive response to radiation. PrxII has been shown to be upregulated in tissues isolated from patients who did not respond to radiation therapy and in radioresistant MCF+FIR3 breast-cancer cells this is associated with breast-cancer radiation sensitivity or resistance.Citation30 This increased PrxII expression confers radioresistance to cancer cells due to its role in protecting cells from radiation-induced cell death. Prxs are important not only as antioxidant proteins, keeping toxic H2O2 and peroxynitrite levels low, but also in such processes as apoptosis, differentiation, and proliferation. Therefore, alteration in PrxII level, determination of PrxII mechanisms of action, and targeting PrxII modulation by chemical or biological means may lead to new strategies that can be developed to improve radiation therapeutic ratios.
We have been using siRNA to modulate the PrxII gene expression level to determine the connection between PrxII expression level and radiosensitivity. We have shown that siRNA can be used to silence protein expression in a gene-specific manner – a process that has found widespread applications in the fields of functional genomics and drug discovery.Citation31 It has become a powerful research tool to target molecules that conventional therapeutics cannot reach. In our studies, the siRNA that inhibited PrxII gene expression partially reversed the radioresistant phenotype of MCF-7 breast cancer. PrxII has a very important role in radioresistance of breast-cancer cells, even though it is not the sole factor responsible for the resistant phenotype. In the study presented here, we showed that inhibition of PrxII gene expression by PrxII-siRNA decreased PSH and GSH levels in MCF+FIR3 radioresistant breast-cancer cells compared with the control cells. Thus, the mechanism of action of PrxII-siRNA sensitization of radioresistant breast-cancer cells involved both GSH and PSH.
It has been shown that processes involving changes in cellular thiol levels can affect Ca2+ signaling pathways that participate in a broad array of physiological responses.Citation34 Previous studies have unequivocally established the key role of calcium as a universal second messenger.Citation32,Citation33 Further, it is becoming increasingly apparent that there are significant interactions between calcium and cellular thiol metabolism, and that these interactions modify a variety of proteins that participate in signaling transduction pathways and other fundamental cellular functions that determine cell life or death. Intracellular Ca2+ homeostasis is regulated by compartmentalization processes involving Ca2+ sequestration in mitochondria and the endoplasmic reticulum and Ca2+ extrusion from the cell by the plasma membrane Ca2+-ATP. When GSH and PSH levels are low, the Ca2+-ATPase pump can be inactivated due to the high level of ROS, which leads to the generation of abnormal calcium signals. This calcium imbalance may be caused by damage to the Ca2+-ATPase pump. Both nuclei and cytoplasmic Ca2+ can regulate phosphorylation and dephosphorylation of proteins and, as a result, modulate signal transduction pathways. Therefore, in the study reported here, inhibition of PrxII gene expression by PrxII-siRNA changed the cellular thiol status and disrupted the normal physiological pathways. As a result, it is very likely that it increased cellular toxicity and rendered MCF+FIR3 breast-cancer cells more sensitive to IR.
Ca2+ levels fluctuate with the change of the Pi levels. Pi has been used as an indicator for Ca2+ level. The increase in Pi levels we observed following IR in PrxII-siRNA radioresistant breast-cancer cells indicated an increased Ca2+ concentration. This is most likely due to the inhibition of the plasma membrane Ca2+ pump in the plasma membrane and the Ca2+ storage deficiency of the mitochondria and endoplasmic reticulum. In control cells, control siRNA treatment did not appear to cause any appreciable inhibition of the plasma membrane Ca2+ pump. The Ca2+ released from the mitochondria and/or endoplasmic reticulum would still be expected to produce at least a transient increase in cytosolic Ca2+ concentration and phosphate level, but it would not last long since the Ca2+ would eventually be extruded through the membrane Ca2+ pump. However, when the plasma membrane Ca2+ pump is damaged, the extrusion of Ca2+ released from the intracellular stores is impaired (PrxII-siRNA + radiation) and a significant increase of phosphate concentration then occurs. The results indicate that PrxII and cellular thiol played a protective role for the plasma membrane against IR-induced damage. The depletion of cellular thiol by PrxII-siRNA correlated well with the radiation response of MCF+FIR3 radioresistant and MCF+FIS4 radiosensitive cells. The results suggest that perturbation of cellular thiol homeostasis by inhibition of PrxII represents a crucial step in modulating cell proliferation and differentiation. Better understanding of the mechanism may also generate novel strategies to overcome the development of MCF-7 breast-cancer resistance and further improve breast-cancer treatment.
Future directions
The results of this study may provide important clinical information on prediction of the behavior and treatment response of MCF-7 radioresistant breast-cancer cells. However, PrxII-siRNA does not completely inhibit PrxII gene expression. The radioresistance phenotype of MCF+FIR3 breast-cancer cells was not fully reversed by targeting the PrxII gene. Some of the more potent subpopulations of radioresistant CSCs may evade therapy and propagate, seeding new tumor growth. This problem poses a great challenge and opportunity for future cancer research. Further investigation will be necessary to elucidate in detail the molecular signaling mechanisms involved in radiation resistance in BCSCs in order to identify alternative targets that can enhance the efficacy of radiotherapy due to its targeted property to specific cancer-related genes. However, PrxII may be potentially important in MCF-7 breast-cancer cells with an increased radioresistant phenotype.
We hope the results presented here will contribute to better understanding of the biochemical and clinical pharmacology of the combination approach of siRNA and therapeutic IR so that this technology can fulfill a strategy to target enzymes in CSCs, the DNA repair pathway, and cellular protection machinery to enhance chemosensitivity.
Disclosure
The authors declare no conflicts of interest in this work.
Acknowledgments
We would like to thank Arnold and Mable Beckman Foundation, Research, Scholarship, Creative Activities Award Program (RSCAAP), California State University Program for Education and Research in Biotechnology (CSUPERB), and National Science Foundation (NSF) #HRD-0802628 for supporting this project. We want to thank Dr Robin Roth and Ms Angela Avatia for proofreading of the paper.
References
- MenaaCLiJJThe role of radiotherapy-resistant stem cells in breast cancer recurrenceBreast Cancer Manage2013228992
- American Cancer SocietyBreast Cancer Facts and Figures 2007–2008Atlanta, GAAmerican Cancer Society Available from: http://www.cancer.org/Research/CancerFactsFigures/BreastCancerFactsFigures/bcff-final-pdfAccessed August 12, 2013
- BonhamVLWarshauer-BakerECollinsFSRace and ethnicity in the genome era: the complexity of the constructsAm Psychol200560191515641917
- PhillipsTMMcBrideWHPajonkFThe response of CD24−/low/CD44+ breast cancer–initiating cells to radiationJ Natl Cancer Inst200698241777178517179479
- DiehnMChoRWLoboNAAssociation of reactive oxygen species levels and radioresistance in cancer stem cellsNature2009458723978078319194462
- WangTTamaeDLeBonTShivelyJEYenYLiJJThe role of peroxiredoxin II in radiation-resistant MCF-7 breast cancer cellsCancer Res20056522103381034616288023
- KowaltowskiAJNettoLEVercesiAEThe thiol-specific antioxidant enzyme prevents mitochondrial permeability transition. Evidence for the participation of reactive oxygen species in this mechanismJ Biol Chem19982732112766127699582302
- KowaltowskiAJVercesiAERheeSGNettoLECatalases and thioredoxin peroxidase protect Saccharomyces cerevisiae against Ca(2+)-induced mitochondrial membrane permeabilization and cell deathFEBS Lett2000473217718210812070
- NettoLEKowaltowskiAJCastilhoRFVercesiAEThiol enzymes protecting mitochondria against oxidative damageMethods Enzymol200234826027011885279
- RheeSGWooHAKilISBaeSHPeroxiredoxin functions as a peroxidase and a regulator and sensor of local peroxidesJ Biol Chem201228774403441022147704
- NettoLESChaeHZKangSWRheeSGStadtmanERRemoval of hydrogen peroxide by thiol-specific antioxidant enzyme (TSA) is involved with its antioxidant properties. TSA possesses thiol peroxidase activityJ Biol Chem19962712615315153218663080
- ButterfieldLHMerinoAGolubSHShauHFrom cytoprotection to tumor suppression: the multifactorial role of peroxiredoxinsAntioxid Redox Signal19991438540211233141
- BiswasSChidaASRahmanIRedox modifications of protein-thiols: emerging roles in cell signalingBiochem Pharmacol200671555156416337153
- ShauHKimATHedrickCCEndogenous natural killer enhancing factor-B increases cellular resistance to oxidative stressesFree Radic Biol Med19972234975078981042
- KimHLeeTHParkESRole of peroxiredoxins in regulating intracellular hydrogen peroxide and hydrogen peroxide-induced apoptosis in thyroid cellsJ Biol Chem200027524182661827010849441
- ChungYMYooYDParkJKKimYTKimHJIncreased expression of peroxiredoxin II confers resistance to cisplatinAnticancer Res2001212A1129113311396151
- SarafianTARajperNGrigorianBKimAShauHCellular antioxidant properties of human natural killer enhancing factor BFree Radic Res19972632812899161849
- WangTHuYDongSCo-activation of ERK, NF-kappaB, and GADD45beta in response to ionizing radiationJ Biol Chem200528013125931260115642734
- WoodZAPooleLBKarplusPAPeroxiredoxin evolution and the regulation of hydrogen peroxide signalingScience2003300561965065312714747
- LushchakVIGlutathione homeostasis and functions: potential targets for medical interventionsJ Amino Acids2012201273683722500213
- WoodZASchröderERobin HarrisJPooleLBStructure, mechanism and regulation of peroxiredoxinsTrends Biochem Sci2003281324012517450
- ZhengMAslundFStorzGActivation of the OxyR transcription factor by reversible disulfide bond formationScience199827913171817219497290
- ChakrabortyCPotentiality of small interfering RNAs (siRNA) as recent therapeutic targets for gene-silencingCurr Drug Targets20078346948217348839
- MittalVImproving the efficiency of RNA interference in mammalsNat Rev Genet20045535536515143318
- WhalleyKBreakthrough for systemic RNAiNat Rev Drug Dis20065373
- Di MonteDBellomoGThorHNicoteraPOrreniusSMenadione-induced cytotoxicity is associated with protein thiol oxidation and alteration in intracellular Ca2+ homeostasisArch Biochem Biophys198423523433506097183
- BellomoGNicoteraPOrreniusSAlterations in intracellular calcium compartmentation following inhibition of calcium efflux from isolated hepatocytesEur J Biochem1984144119236090141
- DuruNFanMCandasDHer2-associated Radioresistance of Breast Cancer Stem Cells Isolated from Her2-Negative Breast Cancer CellsClin Cancer Res2012186634664723091114
- ValentPBonnetDDe MariaRCancer stem cell definitions and terminology: the devil is in the detailsNat Rev Cancer20121211707775
- ParkSChungYMLeeYSAntisense of human peroxiredoxin II enhances radiation-induced cell deathClin Cancer Res20006124915492011156252
- WhalleyKBreakthrough for systemic RNAiNat Rev Drug Dis20065373
- BellomoGJewellSAThorHOrreniusSRegulation of intracellular calcium compartmentation: studies with isolated hepatocytes and t-butyl hydroperoxideProc Natl Acad Sci U S A19827922684268466817329
- SenCKCellular thiols and redox-regulated signal transductionCurr Top Cell Regul20003613010842745
- ClaphamDECalcium signalingCell20071311047105818083096