Abstract
With about 22,000 new cases estimated in 2012 in the US and 15,500 related deaths, ovarian cancer is a heterogeneous and aggressive disease. Even though most of patients are sensitive to chemotherapy treatment following surgery, recurring disease is almost always lethal, and only about 30% of the women affected will be cured. Thanks to a better understanding of the molecular mechanisms underlying ovarian cancer malignancy, new therapeutic options with molecular-targeted agents have become available. This review discusses the rationale behind molecular-targeted therapies and examines how newly identified molecular targets may enhance personalized therapies for ovarian cancer patients.
Introduction to ovarian cancer and current therapies
As knowledge of the complexity and diversity of tumor cells increases, clinical trials attempt to improve the treatment of patients by using molecular-targeted agents. This demarche is based on the assumption that targeting the signaling pathways that a tumor cell depends on, especially if these are driven by the alteration of a protein, might sensitize the tumor cells to treatment without affecting the normal cells. Thus, targeted therapies are emerging for the treatment of several types of tumors characterized by specific and additive genetic aberrations. For example, in lung cancers, patients with mutant EGFRs are treated with EGFR inhibitors, such as erlotinib or gefitinib,Citation1 while breast cancer patients with overexpressed HER2 are treated with agents that inhibit that pathway (eg, trastuzumab or lapatinib).Citation2 Over the past decade, fundamental research has shed light on numerous signaling pathways critical for the growth and the metastasis of ovarian tumor cells. The discovery of BRCA1/2 mutations underlying hereditary ovarian cancers and the use of PARP inhibitors in the clinic prompted the development of targeted therapies for ovarian cancer patients. This review outlines the current state of the emerging molecular-targeted therapies for ovarian cancers, and focuses specifically on molecular-targeted agents that affect some of the hallmarks of cancer: angiogenesis, genomic instability, anti-apoptotic signals, and metabolism.Citation3
Ovarian cancer is the ninth most common cancer, but is the fifth most frequent cause of cancer-related deaths in women. Even though 90% of patients can be cured if they are diagnosed at an early stage (stage 1, when the cancer is limited to the ovaries), over 80% of ovarian cancer patients are diagnosed after the tumor has already metastasized (stage 2 or beyond). The most common type of ovarian cancer (which is observed in approximately 90% of cases) is thought to originate from the epithelial cells covering the ovaries, and is known as ovarian epithelial cancer. Although ovarian cancers are all epithelial in origin, they display four distinct histologies – serous, mucinous, clear cell, and endometrioid – that correlate with distinct gene expression patterns, and with distinct sensitivity to therapies. Also, patients with clear cell histology appear to have a worse prognosis than other histologies.
At early stages (stages 1–2), current therapies include surgery, chemotherapy, and/or radiation therapy. At later stages, debulking surgery will be combined with platinum or taxane-based chemotherapy (intraperitoneal or external) and, potentially, radiation therapy. However, since 80% of patients will relapse after first-line platinum-based or taxane-based chemotherapy, the development of new therapies is needed. A close follow-up of patients with complete clinical remission is commonly done by regularly measuring the levels of serum CA 125, an ovarian-cancer antigen.Citation4–Citation6 Indeed, increases in CA 125 levels (in comparison with the levels obtained at the completion of the first line treatment) allow the early detection of a relapse. Therapeutic options for patients with recurrent disease are extremely limited. For patients with platinum-sensitive disease, treatment with platinum or platinum based combinations may be considered. For other patients, enrollment into a clinical trial might be an option.
Emerging molecular-targeted therapies
Gene amplifications, genetic mutations, and epigenetic abnormalities may lead to aberrant activation of an oncogene or to the loss of function of a tumor suppressor, and, consequently, may promote tumor growth. These genetic aberrations have all been reported in ovarian cancer,Citation7 thus, understanding the biology underlying the development and growth of ovarian cancer may give us the keys for successful targeted therapies.
Angiogenesis and receptor tyrosine kinases
Angiogenesis (the formation of new blood vessels) is a critical parameter for tumor growth and survival as it provides the nutrients and the oxygen necessary to maintain tumor cell biological functions. The best-studied pathway involved in angiogenesis is the vascular endothelial growth factor (VEGF) pathway. The VEGF family consists of seven ligands (VEGF-A to -E, and placental growth factors 1 to 2) and three receptors (VEGFR1 to VEGFR3).Citation8 VEGF signaling is important for normal ovarian physiology and the reproductive cycle,Citation9,Citation10 but ovarian cancer is able to co-opt VEGF signaling. Indeed, retrospective clinical studies and preclinical studies have shown that the VEGF family pathway is activated in ovarian tumors,Citation11 and might indicate a poor prognosis or survival.Citation12,Citation13 At the molecular level, VEGFR activates several signaling pathways such as the PI3K/Akt signaling cascade and the MAP kinase pathway, and therefore promotes tumor growth, survival, and metastasis. Because VEGFRs are expressed and functional in ovarian cancer cells,Citation14 anti-angiogenic therapies may also have a direct anti-tumor effect.Citation15 Direct inhibitors of VEGF-A using monoclonal antibodies (bevacizumab, VEGFTrap) as well as multiple small molecules inhibiting VEGFR have both been broadly developed,Citation8 although most of these molecules inhibit other receptor tyrosine kinases.Citation16,Citation17 For example, vandetanib inhibits VEGFR, RET, and EGFR, while sorafenib is a VEGFR, PDGFR, and c-Kit inhibitor, and pazopanib inhibits VEGFR and PDGFR.
Clinical results of two highly anticipated phase III trials using the VEGF monoclonal antibody bevacizumab (Avastin™; Genentech/Roche, South San Francisco, CA) have recently been published.Citation18,Citation19 Avastin slowed tumor growth, but no significant difference in overall survival was observed in one of the studies. Other clinical trials targeting VEGFR using small molecules such as pazopanib, cediranib, sorafenib, and vandetanib were performed or are ongoing. A phase II trial with pazopanib in patients with recurrent disease appears to be promising,Citation20 but a phase II trial combining sorafenib with topotecan presented high toxicity and poor clinical activity,Citation21 and a phase II trial found that vandetanib had no clinical activity as monotherapy for recurrent ovarian cancers.Citation22 There may be several reasons for this variability in effect. One obvious concern is the poor specificity of VEGF receptor kinase inhibitors,Citation16,Citation17 which suggests that while targeting the vasculature of ovarian cancer is attractive, an understanding of the broad molecular effects of each receptor tyrosine kinase inhibitor (RTKI) will be necessary to better design these therapies.
The targeting of angiogenesis through alternative pathways mediated by RTKs other than VEGFR has been well studied in both preclinical and clinical studies of ovarian cancer. EGFR, Src, and Met have overlapping functions in the activation of signaling pathways involved in angiogenesis, cell growth, survival, and metastasis.
The EGFR receptor tyrosine kinase family has four members: EGFR, ErB2/HER2, ErB3/HER3, and ErB4/HER4. Following ligand binding, EGFR dimerizes and activates signaling pathways such as PI3K/Akt and MAPK. Overexpression of EGFR has been observed in ovarian cancer and its nuclear localization has been linked to poor prognosis. The effect of EGFR inhibitors on ovarian cancer has been clinically investigated. Pertuzumab, a monoclonal antibody against EGFR, has shown encouraging results when combined with chemotherapy.Citation23 However, other EGFR inhibitors such as gefitinib or trastuzumab presented variable clinical activities,Citation24,Citation25 suggesting that better patient selection with the development of new prognostic biomarkers might be needed.
Expression and activation of the nonreceptor tyrosine kinase Src leads to tumor cell growth, survival, and metastasis, and is an indicator of poor prognosis in ovarian tumors. The Src kinase family has nine members: Src, Fyn, Yes, Lyn, Lck, Fgr, Blk, Hck, and Yrk. Both Src and Yes have been shown to be overexpressed and activated in late stage ovarian cancer,Citation26 and are key mediators of various RTKs, such as EGFR, Met, VEGFR, or HER2. Src activation promotes angiogenesis and invasion by supporting VEGF-A expressionCitation27–Citation29 and inhibiting the expression of anti-angiogenic factors mediated by TGFβ1.Citation30 Src activation has also been linked to platinum-drug resistance.Citation31 This suggests that inhibition of Src combined with paclitaxel or with an anti-angiogenic agent might have a therapeutic value by decreasing the development of resistance to these therapies. A phase II and III clinical trial using the Src inhibitor saracatinib (AZD0530) combined with paclitaxel is ongoing in platinum resistant ovarian cancer patients.
Since the first report of Met being an oncogene was published in 1984,Citation32 several solid tumors have been shown to be driven by Met aberrant activation and/or expression. Papillary type one kidney tumors and ovarian cancers are two examples out of many.Citation33–Citation35 Aberrant activation of Met can be due to overexpression of its endogenous ligand, the hepatocyte growth factor/scatter factor (HGF/SF), as well as point mutations of the MET gene, or activation of other receptors such as EGFR,Citation36,Citation37 semaphorin 4D receptor,Citation38 or α5-integrinCitation39 that can all activate Met by heterodimerization. This indicates that Met is involved in the crosstalk of multiple signaling pathways and plays an important role in tumor growth and metastasis.Citation37 Met and its ligand HGF/SF also play an important role in angiogenesis. HGF/SF was described in 1992 as a “potent angiogenic factor which stimulates endothelial cells motility and growth.”Citation40 Since then, Met has been shown to regulate VEGF-A signaling.Citation37,Citation41,Citation42 In ovarian cancer, several pre-clinical studies have identified Met as a relevant therapeutic target due in part to its role in invasionCitation43 and angiogenesis,Citation37 which suggests that combining Met inhibition with anti-angiogenic therapies could be beneficial for ovarian cancer patients. Since Met is also important in the development of resistance to therapies targeting EGFRCitation44 it would be interesting to examine whether dual EGFR/Met inhibition improves the effect of EGFR inhibitors in the clinic.
Genomic instability and BRCA1/BRCA2
Patients with mutations in one of the tumor-suppressor genes BRCA1 or BRCA2 are more likely to develop breast and ovarian cancers following the loss of the remaining wild-type allele (LOH). BRCA1 and BRCA2 encode for proteins that maintain the integrity of the genome by regulating the DNA damage response and repair. Although mutations in BRCA1 or BRCA2 lead to similar diseases, the proteins they code for have different functions with BRCA1 involved in both the DNA damage response and DNA repair, whereas BRCA2 is involved only in DNA repair; however, both are critical for homologous recombination (HR).Citation45 When a double-strand break occurs in proliferating cells, HR will repair the DNA with high fidelity. When BRCA1 or BRCA2 are mutated and HR is compromised, the overall repair capacity of the cell is greatly reduced, and less reliable repair pathways such as nonhomologous end-joining (NHEJ) will be used, leading to increased genomic instability. It has been shown that ovarian cancer patients with BRCA mutations may be more sensitive to platinum-based chemotherapy and may have better outcomes than patients without BRCA mutations.Citation46,Citation47 PARP proteins are also involved in DNA repair and thus BRCA mutated cells are highly sensitive to PARP inhibition. Alterations of other members of the HR pathway such as ATM, ATR, CHK1, or CHK2 also sensitize the cells to PARP inhibitors. Since mutations of ATM and CHK2 are common in cancers with deficient BRCA and further increase genome instability,Citation45,Citation48,Citation49 one can predict that cells with several mutations in the HR pathway will have a marked sensitivity towards PARP inhibitors. Several PARP inhibitors are currently used in the clinic for patients with BRCA mutations or methylation. In a recent randomized phase II multicenter study, the efficacy of the PARP inhibitor olaparib was compared with pegylated liposomal doxorubicin in patients with BRCA1/2 mutations and recurrent ovarian cancer, but no significant difference was reported.Citation50 The existence of secondary somatic mutations able to restore BRCA functions has been proposed to explain these results.Citation51,Citation52
Another way to selectively target patients with BRCA loss of function could be through the regulation of epigenetic modifications. Posttranslational modification of histones by methylation and acetylation regulates the access of a transcription factor to the DNA, so this plays a prominent role in controlling gene expression. In tumors, it is common to observe aberrant DNA methylation that silences tumor-suppressor gene expression. In ovarian cancer the BRCA1 promoter has been shown to be hypermethylated in more than 30% of tumors.Citation53 In contrast, acetylation of histones on lysine residues by histone acetyltransferases (HAT) allows the active transcription of genes. Acetyl groups are removed by histone deacetylases (HDACs), silencing gene expression. Thus, while the mechanism of action of HDAC inhibitors is not fully understood, they may allow the reactivation of silenced genes. Several promising preclinical studies in ovarian tumor cells have been performed using HDAC inhibitors either alone or in combination with other agents.Citation54–Citation57 Because inhibition of HDACs has been shown to reverse epigenetic silencing,Citation58 it is reasonable to speculate that combining HDAC inhibitors with paclitaxel and/or cisplatin, or with PARP inhibitors, might be of therapeutic value, especially if targeted to patients with silenced BRCA genes. A phase III clinical trial for cisplatin-resistant ovarian cancer with topotecan alone or combined with the epigenetic agents hydralazine and magnesium valproate is currently ongoing at the National Institute of Cancerologia (Mexico).
Anti-apoptosis and cell survival: the PI3K/Akt pathway
The PI3K/Akt signaling pathway plays a prominent role in the growth and survival of numerous cancers, including ovarian cancer.Citation59–Citation61 The three classes of the lipid kinases PI3K are made up of a regulatory (p85) and a catalytic (p110) subunit, with several isoforms for each. In cancer, inappropriately activated PI3K induces the phosphorylation of phosphatidylinositols, especially phosphatidylinositol-4,5-biphosphate (PIP2), to produce phosphatidylinositol-3,4,5-trisphosphate (PIP3). PI3K activation is counterbalanced by PTEN, a 3′-phosphatase that transforms PIP3 into PIP2. The second messenger, PIP3, recruits and promotes the activation of Akt, which initiates a signaling cascade leading to anti-apoptotic signals, and tumor cell growth and survival.Citation59 In ovarian cancer, genetic mutations have been found on PTEN*, PIK3CA (p110α)* (*from the Catalogue Of Somatic Mutations in Cancer) PIK3R1 (p85),Citation62 and AKT2.Citation63,Citation64 All of these mutations can lead to aberrant and constitutive activation of the PI3K/Akt signaling cascade, and to the subsequent activation of mTOR and NFkB, two targets of the Akt pathway. Because aberrant constitutive activation of the PI3K/Akt pathway promotes tumor survival and chemoresistance, small inhibitors targeting this signaling cascade may have a therapeutic value. Several clinical trials targeting the PI3K/Akt pathway are ongoing, and pan-PI3K inhibitors are currently in phase I clinical trials. For example, XL147 is in trials in combination with paclitaxel and carboplatin, and BKM120 is used as a single agent for patients with specific PIK3CA mutations. The pan-Akt inhibitor MK-2206 is in phase II trial. Small molecules targeting downstream targets of Akt such as mTOR inhibitors are also used in combination with carboplatin and taxol/paclitaxel in clinical trials. Ridaforolimus is in phase I trial, temsirolimus is in phase II trial, and the mTOR inhibitor RAD001 combined with avastin is also in phase II clinical trials. As well as this, even though MAPK mutations were suspected to induce resistance to PI3K/Akt/mTOR inhibitors, it has recently been demonstrated that a subset of patients with both PIK3CA and MAPK mutations responded to PI3K/Akt/mTor targeted therapies.Citation65
Promising preclinical data linked to the aberrant activation of the PI3K/Akt signaling cascade identify potential new therapeutic targets. For example, loss of tumor suppressors such as PTEN, DNA damage, or genetic alterations can also lead to the aberrant activation of the nuclear factor-κB (NF-κB). In cancer cells, the transcription factor NF-κB plays a complex role that promotes angiogenesis, inflammation, and metastasis.Citation66 Preclinical data have shown that the NF-κB pathway is critical for ovarian cancer cellsCitation67 and the development of selective and direct NF-κB inhibitors may have therapeutic value for ovarian cancer patients. Moreover, humoral hypercalcemia of malignancy (HHM) is mediated by the secretion of parathyroid hormone-related peptide (PTHrP) and has been associated with gynecologic neoplasms, including ovarian cancers.Citation68 Patients with HHM will present with hypercalcemia, low parathyroid hormone (PTH), and high PTHrP serum levels. At the molecular level, PTHrP binds to the PTH/PTHrP receptors and activates the PI3K/Akt/NF-κB pathway,Citation69 which promotes tumor growth and metastasis. As a result, ovarian cancer patients with HHM might particularly benefit from a therapy targeting the PI3K/Akt signaling cascade.
Besides its role in promoting anti-apoptosis and cell survival, the PI3K/Akt pathway, especially mTOR, is a key node in regulating cellular metabolism. Indeed, activation of Akt increases glycolysis,Citation70 decreases β-oxidation by reducing the expression of carnitine palmitoyltransferase 1A (CPT1A),Citation71 upregulates the fatty acid synthase (FASN),Citation72,Citation73 and activates mTOR. Thus, by promoting glycolysis and anabolic reactions, the PI3K/Akt pathway promotes an anabolic metabolism shift that might favor the growth and spread of tumor cells.
Metabolism of ovarian cancer cells: from HIF1α to the mitochondria
It is now broadly accepted that metabolic reprogramming of tumor cells, either as a tumor initiator or as a consequence of tumor growth, provides a growth advantage to tumor cells. The mechanisms underlying this reprogramming are, however, not fully understood.
The hypoxia inducible factor HIF1α is critical to maintain the energy production of a cell in the metabolic shift that occurs under hypoxia.Citation74 In normal tissues, oxygen is consumed by the cells to support mitochondrial function and produce energy by oxidative phosphorylation. Under hypoxia, oxidative phosphorylation is impaired because there is little or no oxygen to serve as an electron acceptor at the end of the electron transport chain, which leads to a decrease in ATP production. A metabolic shift needs to occur to compensate for this lack of energy, and this metabolic adaptation can be driven by HIF1α. In the absence of oxygen, HIF1α is stabilized and translocates into the nucleus to transcribe the genes involved in glucose metabolismCitation74 such as Glut1, lactate dehydrogenase A (LDHA), or pyruvate kinase (PK) M2 (which is also a coactivator of HIF1α).Citation75 By activating these genes, HIF1α promotes glycolysis, a less efficient yet reliable way to produce energy under hypoxic conditions.Citation74 In tumors where HIF1α is expressed, this metabolic shift occurs even in the presence of oxygen and is known as aerobic glycolysis, or the Warburg effect.Citation76
HIF1α is overexpressed in late stage ovarian cancer,Citation77 but, to date, its impact on chemotherapy response or prognosis is unclear. That said, low expression of the glucose transporter Glut1, which is its downstream target, correlates with a longer disease-free survival, suggesting that HIF1α could be a therapeutic target. Since VEGF-A is also a transcriptional target of HIF1α, inhibition of HIF1α may simultaneously deprive tumor cells of their energy by inhibiting glucose metabolism and angiogenesis. Although no direct HIF1α inhibitors are currently available in the clinic, several small molecules affecting alternative pathways have been shown to decrease HIF1α expression and activity. Topoisomerase inhibitors,Citation78 HSP90 inhibitors,Citation79 mTOR inhibitors,Citation80 and EGFR inhibitorsCitation81 have all shown potent anti-HIF1α activity in preclinical studies, and, with the exception of HSP90 inhibitors, are currently used at different phases in the clinic for the treatment of ovarian cancer patients. HIF1α expression may also have a prognostic or diagnostic value in those studies.
Another possible metabolic adaptation promoting rapid growth of tumors is their parasitic use of energetic resources from surrounding tissues. Recently, Nieman et al have demonstrated that adipocytes can be an energy reservoir for ovarian tumor cells.Citation82 Surrounding adipocytes promote migration and invasion by transferring fatty acids to the tumor cells, which will then generate ATP via mitochondrial β-oxidation. Thus, any agents that can prevent tumor cells receiving energy from their supporting host cells could have a therapeutic effect, and this includes AMPK activators (eg, metformin) or lipid synthase inhibitors. Since Akt is known to decrease mitochondrial β-oxidation, PI3k/Akt inhibitors might prevent the parasitic usage of energetic resources by ovarian cancer cells and might have a therapeutic value. Moreover, the small molecule elesclomol (STA4783), which targets mitochondrial function and requires a functional electron transport chain to show a cytotoxic effect,Citation83,Citation84 is currently in phase II clinical trial for ovarian cancers. This agent may be more selective for “parasitic” tumor cells that preserve functional mitochondria, while not affecting surrounding adipocytes since these have a shifted metabolism with increased glycolysis and lipolysis.
Conclusion
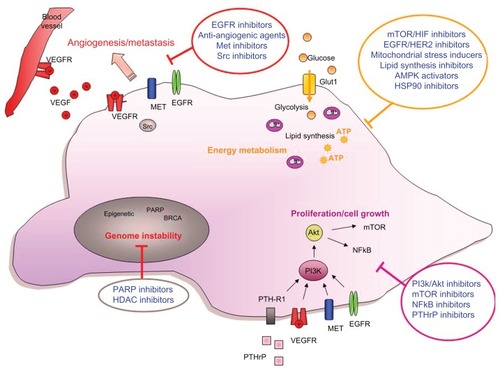
Molecular characterization of ovarian cancer is already allowing for improvements in the design of targeted therapies. Over 72,000 scientific papers related to ovarian cancer have been published on PubMed, and numerous gene mutations have been identified by The Cancer Genome Atlas (http://cancergenome.nih.gov). This allows for the development of multiple therapeutic approaches with the potential to treat ovarian cancer. Several emerging targeted therapies have been highlighted in this review. They target multiple aspects of the hallmarks of a cancer cell () and expand the current treatment options for ovarian cancer patients.
Figure 1 Targeting ovarian cancer cells.
Abbreviations: PTHrP, parathyroid hormone related protein; VEGF, vascular endothelial growth factor; Glut1, glucose transporter 1; NFkB, nuclear factor-kB; EGFR, epidermal growth factor receptor; PI3K, phosphoinositide-3-kinase; PARP, poly (ADP-ribose) polymerase; HIF, hypoxia inducible factor.
The complexity of signaling cascades, the numerous resistance mechanisms, and the lack of specificity of certain small molecules all make it difficult to predict which therapy will be successful, or identify the appropriate patient populations. An in-depth understanding of the molecular effects that a small molecule may have toward diverse types of tumors, and how this might lead to resistance, will certainly be an advantage. The use of new targeted agents will be improved by the development of multiple biomarkers to identify which patients will benefit or be harmed by a particular treatment, and to monitor the efficacy of treatments. Understanding both the mechanisms of action and the toxicities of therapeutic agents will ultimately enhance the quality of patient care and the quality of life of ovarian cancer patients.
Other challenges that were not discussed in this review also remain. Understanding the roles played by stem cellsCitation85 or microRNACitation86 in the growth of ovarian tumors, improving the delivery of therapies using nanotechnologies,Citation87 as well as studying the prognostic value of surgical resection of tumor necrotic tissues following therapy, or assessing the prognostic value of minimal residual disease for ovarian cancer patients, are a few examples of these challenges. A better understanding of these will improve the treatment and standard of care for patients with advanced ovarian cancer. For example, a recent prospective multicenter study showed that patients with complete cytoreduction have a better outcome than patients with residual minimal disease,Citation88 a finding that will certainly have a significant impact on the care of ovarian cancer patients.
Disclosure
The author reports no conflicts of interest in this work. This review was prepared by the author in her personal capacity. Ongoing clinical trials were found using the http://www.clinicaltrials.gov and http://www.clinicaltrials.com websites, as well as Internet searches. The opinions expressed in this article are the author’s own and do not necessarily reflect the views of any institutions and/or governments.
References
- RukazenkovYSpeakeGMarshallGEpidermal growth factor receptor tyrosine kinase inhibitors: similar but different?Anticancer Drugs2009201085686619657272
- BaselgaJBradburyIEidtmannHLapatinib with trastuzumab for HER2-positive early breast cancer (NeoALTTO): a randomised, open-label, multicentre, phase 3 trialLancet2012379981663364022257673
- HanahanDWeinbergRAHallmarks of cancer: the next generationCell2011144564667421376230
- NiloffJMBastRCJrSchaetzlEMKnappRCPredictive value of CA 125 antigen levels in second-look procedures for ovarian cancerAm J Obstet Gynecol198515179819863157319
- NiloffJMKnappRCSchaetzlEReynoldsCBastRCJrCA 125 antigen levels in obstetric and gynecologic patientsObstet Gynecol19846457037076208522
- NiloffJMKnappRCLavinPTThe CA 125 assay as a predictor of clinical recurrence in epithelial ovarian cancerAm J Obstet Gynecol1986155156603460341
- BastRCJrHennessyBMillsGBThe biology of ovarian cancer: new opportunities for translationNat Rev Cancer20099641542819461667
- FerraraNKerbelRSAngiogenesis as a therapeutic targetNature2005438707096797416355214
- FraserHMWilsonHMorrisKDSwanstonIWiegandSJVascular endothelial growth factor Trap suppresses ovarian function at all stages of the luteal phase in the macaqueJ Clin Endocrinol Metab200590105811581816046580
- FraserHMDuncanWCVascular morphogenesis in the primate ovaryAngiogenesis20058210111616240058
- BossEAMassugerLFThomasCMGeurts-MoespotABoonstraHSweepCGVascular endothelial growth factor in ovarian cyst fluidCancer200191237137711180084
- ShenGHGhazizadehMKawanamiOPrognostic significance of vascular endothelial growth factor expression in human ovarian carcinomaBr J Cancer200083219620310901370
- HeflerLAMusteaAKonsgenDVascular endothelial growth factor gene polymorphisms are associated with prognosis in ovarian cancerClin Cancer Res200713389890117289883
- ChenHYeDXieXChenBLuWVEGF, VEGFRs expressions and activated STATs in ovarian epithelial carcinomaGynecol Oncol200494363063515350351
- KumaranGCJaysonGCClampARAntiangiogenic drugs in ovarian cancerBr J Cancer200910011719002176
- DavisMIHuntJPHerrgardSComprehensive analysis of kinase inhibitor selectivityNat Biotechnol201129111046105122037378
- KaramanMWHerrgardSTreiberDKA quantitative analysis of kinase inhibitor selectivityNat Biotechnol200826112713218183025
- PerrenTJSwartAMPfistererJA phase 3 trial of bevacizumab in ovarian cancerN Engl J Med2011365262484249622204725
- BurgerRABradyMFBookmanMAIncorporation of bevacizumab in the primary treatment of ovarian cancerN Engl J Med2011365262473248322204724
- FriedlanderMHancockKCRischinDA Phase II, open-label study evaluating pazopanib in patients with recurrent ovarian cancerGynecol Oncol20101191323720584542
- RamasubbaiahRPerkinsSMSchilderJSorafenib in combination with weekly topotecan in recurrent ovarian cancer, a phase I/II study of the Hoosier Oncology GroupGynecol Oncol2011123349950421955480
- AnnunziataCMWalkerAJMinasianLVandetanib, designed to inhibit VEGFR2 and EGFR signaling, had no clinical activity as monotherapy for recurrent ovarian cancer and no detectable modulation of VEGFR2Clin Cancer Res201016266467220068097
- MakhijaSAmlerLCGlennDClinical activity of gemcitabine plus pertuzumab in platinum-resistant ovarian cancer, fallopian tube cancer, or primary peritoneal cancerJ Clin Oncol20102871215122319901115
- MurphyMStordalBErlotinib or gefitinib for the treatment of relapsed platinum pretreated non-small cell lung cancer and ovarian cancer: a systematic reviewDrug Resist Updat201114317719021435938
- LedermannJARajaFATargeted trials in ovarian cancerGynecol Oncol2010119115115620591473
- WienerJRWindhamTCEstrellaVCActivated SRC protein tyrosine kinase is overexpressed in late-stage human ovarian cancersGynecol Oncol2003881737912504632
- WeisSCuiJBarnesLChereshDEndothelial barrier disruption by VEGF-mediated Src activity potentiates tumor cell extravasation and metastasisJ Cell Biol2004167222322915504909
- MukhopadhyayDTsiokasLSukhatmeVPWild-type p53 and v-Src exert opposing influences on human vascular endothelial growth factor gene expressionCancer Res19955524616161658521408
- MukhopadhyayDTsiokasLZhouXMFosterDBruggeJSSukhatmeVPHypoxic induction of human vascular endothelial growth factor expression through c-Src activationNature199537565325775817540725
- WakaharaKKobayashiHYagyuTTransforming growth factor-beta1-dependent activation of Smad2/3 and up-regulation of PAI-1 expression is negatively regulated by Src in SKOV-3 human ovarian cancer cellsJ Cell Biochem200493343745315372629
- LeXFBastRCJrSrc family kinases and paclitaxel sensitivityCancer Biol Ther201112426026921646863
- CooperCSParkMBlairDGMolecular cloning of a new transforming gene from a chemically transformed human cell lineNature1984311598129336590967
- NataliPGPratMNicotraMROverexpression of the met/HGF receptor in renal cell carcinomasInt J Cancer19966932122178682590
- Di RenzoMFOliveroMKatsarosDOverexpression of the Met/HGF receptor in ovarian cancerInt J Cancer19945856586628077049
- SawadaKRadjabiARShinomiyaNc-Met overexpression is a prognostic factor in ovarian cancer and an effective target for inhibition of peritoneal dissemination and invasionCancer Res20076741670167917308108
- TrusolinoLBertottiAComoglioPMMET signalling: principles and functions in development, organ regeneration and cancerNat Rev Mol Cell Biol2010111283484821102609
- GherardiEBirchmeierWBirchmeierCVandeWGTargeting MET in cancer: rationale and progressNat Rev Cancer20121228910322270953
- GiordanoSCorsoSConrottoPThe semaphorin 4D receptor controls invasive growth by coupling with MetNat Cell Biol20024972072412198496
- MitraAKSawadaKTiwariPMuiKGwinKLengyelELigand-independent activation of c-Met by fibronectin and alpha(5)beta(1)-integrin regulates ovarian cancer invasion and metastasisOncogene201130131566157621119598
- BussolinoFDi RenzoMFZicheMHepatocyte growth factor is a potent angiogenic factor which stimulates endothelial cell motility and growthJ Cell Biol199211936296411383237
- ZhangYWSuYVolpertOVVande WoudeGFHepatocyte growth factor/scatter factor mediates angiogenesis through positive VEGF and negative thrombospondin 1 regulationProc Natl Acad Sci U S A200310022127181272314555767
- SulpiceEDingSMuscatelli-GrouxBCross-talk between the VEGF-A and HGF signalling pathways in endothelial cellsBiol Cell2009101952553919281453
- BirchmeierCBirchmeierWGherardiEVande WoudeGFMet, metastasis, motility and moreNat Rev Mol Cell Biol200341291592514685170
- EngelmanJAZejnullahuKMitsudomiTMET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signalingScience200731658271039104317463250
- RoyRChunJPowellSNBRCA1 and BRCA2: different roles in a common pathway of genome protectionNat Rev Cancer2012121687822193408
- CassIBaldwinRLVarkeyTMoslehiRNarodSAKarlanBYImproved survival in women with BRCA-associated ovarian carcinomaCancer20039792187219512712470
- TanDSRothermundtCThomasKBRCAness” syndrome in ovarian cancer: a case-control study describing the clinical features and outcome of patients with epithelial ovarian cancer associated with BRCA1 and BRCA2 mutationsJ Clin Oncol200826345530553618955455
- TommiskaJBartkovaJHeinonenMThe DNA damage signalling kinase ATM is aberrantly reduced or lost in BRCA1/BRCA2-deficient and ER/PR/ERBB2-triple-negative breast cancerOncogene2008272501250617982490
- SullivanAYuilleMRepellinCConcomitant inactivation of p53 and Chk2 in breast cancerOncogene20022191316132411857075
- KayeSBLubinskiJMatulonisUPhase II, open-label, randomized, multicenter study comparing the efficacy and safety of olaparib, a poly (ADP-ribose) polymerase inhibitor, and pegylated liposomal doxorubicin in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancerJ Clin Oncol201230437237922203755
- NorquistBWurzKAPennilCCSecondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomasJ Clin Oncol201129223008301521709188
- KonstantinopoulosPACannistraSAComparing poly (ADP-ribose) polymerase inhibitors with standard chemotherapy in BRCA-mutated, recurrent ovarian cancer: lessons learned from a negative trialJ Clin Oncol201230434735022203759
- BaldwinRLNemethETranHBRCA1 promoter region hypermethylation in ovarian carcinoma: a population-based studyCancer Res200060195329533311034065
- ChobanianNHGreenbergVLGassJMDesimoneCPVan NagellJRZimmerSGHistone deacetylase inhibitors enhance paclitaxel-induced cell death in ovarian cancer cell lines independent of p53 statusAnticancer Res2004242B53954515160991
- SonnemannJHulsISiglerMHistone deacetylase inhibitors and aspirin interact synergistically to induce cell death in ovarian cancer cellsOncol Rep200820121922418575740
- HoffmanMABlessingJANunezERA phase II trial of CI-958 in recurrent platinum-sensitive ovarian carcinoma: a Gynecologic Oncology Group studyGynecol Oncol200181343343511371134
- HoffmanMABlessingJAMorganMPhase II trial of CI-958 in recurrent platinum-refractory ovarian carcinoma. A Gynecologic Oncology Group StudyGynecol Oncol200079346346511104620
- MarksPAXuWSHistone deacetylase inhibitors: Potential in cancer therapyJ Cell Biochem2009107460060819459166
- EngelmanJATargeting PI3K signalling in cancer: opportunities, challenges and limitationsNat Rev Cancer20099855056219629070
- AltomareDAWangHQSkeleKLAKT and mTOR phosphorylation is frequently detected in ovarian cancer and can be targeted to disrupt ovarian tumor cell growthOncogene200423345853585715208673
- HanrahanAJSchultzNWestfalMLGenomic complexity and AKT dependence in serous ovarian cancerCancer Discov201221566722328975
- PhilpAJCampbellIGLeetCThe phosphatidylinositol 3′-kinase p85alpha gene is an oncogene in human ovarian and colon tumorsCancer Res200161207426742911606375
- BellacosaAdeFDGodwinAKMolecular alterations of the AKT2 oncogene in ovarian and breast carcinomasInt J Cancer19956442802857657393
- ChengJQGodwinAKBellacosaAAKT2, a putative oncogene encoding a member of a subfamily of protein-serine/threonine kinases, is amplified in human ovarian carcinomasProc Natl Acad Sci U S A19928919926792711409633
- JankuFWhelerJJWestinSNPI3K/AKT/mTOR Inhibitors in Patients With Breast and Gynecologic Malignancies Harboring PIK3CA MutationsJ Clin Oncol201230877778222271473
- PerkinsNDThe diverse and complex roles of NF-kappaB subunits in cancerNat Rev Cancer201212212113222257950
- WhiteKLRiderDNKalliKRKnutsonKLJarvikGPGoodeELGenomics of the NF-kappaB signaling pathway: hypothesized role in ovarian cancerCancer Causes Control201122578580121359843
- SavvariPPeitsidisPAlevizakiMDimopoulosMAAntsaklisAPapadimitriouCAParaneoplastic humorally mediated hypercalcemia induced by parathyroid hormone-related protein in gynecologic malignancies: a systematic reviewOnkologie2009328–951752319745599
- AgouniASourbierCDanilinSParathyroid hormone-related protein induces cell survival in human renal cell carcinoma through the PI3K Akt pathway: evidence for a critical role for integrin-linked kinase and nuclear factor kappa BCarcinogenesis20072891893190117468516
- ShawRJCantleyLCDecoding key nodes in the metabolism of cancer cells: sugar and spice and all things niceF1000 Biol Rep20124222242042
- DeberardinisRJLumJJThompsonCBPhosphatidylinositol 3-kinase-dependent modulation of carnitine palmitoyltransferase 1A expression regulates lipid metabolism during hematopoietic cell growthJ Biol Chem200628149373723738017030509
- WangHQAltomareDASkeleKLPositive feedback regulation between AKT activation and fatty acid synthase expression in ovarian carcinoma cellsOncogene200524223574358215806173
- Brahimi-HornMCBellotGPouyssegurJHypoxia and energetic tumour metabolismCurr Opin Genet Dev2011211677221074987
- DenkoNCHypoxia, HIF1 and glucose metabolism in the solid tumourNat Rev Cancer20088970571319143055
- LuoWHuHChangRPyruvate kinase M2 is a PHD3- stimulated coactivator for hypoxia-inducible factor 1Cell2011145573274421620138
- WarburgOOn respiratory impairment in cancer cellsScience1956124321526927013351639
- SeeberLMHorreeNVooijsMAThe role of hypoxia inducible factor-1alpha in gynecological cancerCrit Rev Oncol Hematol201178317318420627616
- RapisardaAUranchimegBSordetOPommierYShoemakerRHMelilloGTopoisomerase I-mediated inhibition of hypoxia-inducible factor 1: mechanism and therapeutic implicationsCancer Res20046441475148214983893
- IsaacsJSJungYJMimnaughEGMartinezACuttittaFNeckersLMHsp90 regulates a von Hippel Lindau-independent hypoxia-inducible factor-1 alpha-degradative pathwayJ Biol Chem200227733299362994412052835
- HudsonCCLiuMChiangGGRegulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycinMol Cell Biol200222207004701412242281
- PoreNJiangZGuptaACernigliaGKaoGDMaityAEGFR tyrosine kinase inhibitors decrease VEGF expression by both hypoxia-inducible factor (HIF)-1-independent and HIF-1-dependent mechanismsCancer Res20066663197320416540671
- NiemanKMKennyHAPenickaCVAdipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growthNat Med201117111498150322037646
- BlackmanRKCheung-OngKGebbiaMMitochondrial electron transport is the cellular target of the oncology drug elesclomolPLoS One201271e2979822253786
- FuldaSGalluzziLKroemerGTargeting mitochondria for cancer therapyNat Rev Drug Discov20109644746420467424
- LimMCSongYJSeoSSYooCWKangSParkSYResidual cancer stem cells after interval cytoreductive surgery following neoadjuvant chemotherapy could result in poor treatment outcomes for ovarian cancerOnkologie201033632433020523098
- DahiyaNMorinPJMicroRNAs in ovarian carcinomasEndocr Relat Cancer2010171F77F8919903743
- SchroederAHellerDAWinslowMMTreating metastatic cancer with nanotechnologyNat Rev Cancer2012121395022193407
- PolterauerSVergoteIConcinNPrognostic value of residual tumor size in patients with epithelial ovarian cancer FIGO stages IIA–IV: analysis of the OVCAD dataInt J Gynecol Cancer201222338038522266934