Abstract
Lung cancer treatment has rapidly changed in the last few years thanks to novel insights into cancer biology. Several biomarkers and signaling pathways have been recognized as conceivable targets for treatment, and among them is the mesenchymal–epithelial transition/hepatocyte growth factor (c-MET/HGF) axis. Alterations in the c-MET gene and aberrations of MET and HGF expression impact on lung cancer prognosis and are involved in resistance to epidermal growth factor receptor (EGFR) inhibitors in non-small cell lung cancer (NSCLC) patients harboring activating EGFR mutations. Several anti-MET and anti-HGF strategies are currently under investigation, including monoclonal antibodies. Ficlatuzumab is a monoclonal antibody directed against HGF that is currently under investigation in NSCLC. The aim of the present review is to critically review available data on HGF and ficlatuzumab in NSCLC.
Introduction
Lung cancer is a big killer in oncology, accounting for 1.3 million deaths per year worldwide.Citation1 This disease includes two major histologic categories: small-cell lung cancer (SCLC, 15%–20% of cases) and non-small cell lung cancer (NSCLC, 80%–85% of cases) including adenocarcinoma, squamous cell carcinoma and large cell carcinoma. Beyond histologic aspects, lung cancer differs by the molecular aberration at the base of its pathogenesis and sustenance. Several oncogenic alterations in the genetic code and protein expression have so far been identified as conceivable targets for treatment. These molecular aberrations define subsets of patients with specific prognosis and outcome following treatment. Epidermal growth factor receptor (EGFR) gene mutations can be detected in 10%–15% of Caucasians and in up to 40% of Asian NSCLC patients. Soon after their discovery in 2004, they were recognized as the principal biomarker in lung adenocarcinoma predicting response to treatment with the EGFR tyrosine kinase inhibitors (TKI).Citation2 Seven phase III trials including thousands of patients treated with gefitinib, erlotinib, or afatinib, clearly demonstrated that EGFR TKI are the best option as first-line therapy for EGFR mutated NSCLC.Citation3–Citation9 Disease control can be reached in up to 90% of mutant individuals, but none of them can be definitively cured and progression of disease inevitably occurs. Moreover, a consistent proportion of patients show primary resistance to EGFR inhibitors, even in the presence of EGFR activating mutations. Resistance is usually determined by secondary genomic alterations in the target kinase altering the physical or biochemical properties of the receptor and by the activation of collateral pathways. In 50% of cases a secondary gatekeeper mutation in the EGFR gene (T790M, D761Y) is responsible for acquired resistance.Citation11–Citation13 An additional 20% of refractory patients harbor overexpression of another tyrosine kinase receptor, the mesenchymal–epithelial transition (MET) receptor, which allows inhibition of the EGFR pathway to be bypassed.Citation14,Citation15 Some preclinical studies described a correlation between EGFR TKI resistance and overexpression of the c-MET ligand, hepatocyte growth factor (HGF).Citation16 Several strategies to overcome resistance to EGFR TKI are being explored in preclinical and clinical trials. In case of a secondary mutation, irreversible TKI,Citation9 heat shock protein 90 inhibitors,Citation17 or combined treatment with anti-EGFR antibodiesCitation18 are under evaluation. Several MET inhibitors have so far been developed including monoclonal antibodies (ornatuzumab) and small molecule inhibitors (crizotinib, foretinib, cabozantinib, GCD265, tivantinib).Citation19–Citation24 Another possible strategy under evaluation is the blockade of HGF by competitive antagonists (NK4) or specific antibodies (AMG102/rilotumumab, AV-299/ficlatuzumab).Citation25,Citation26 In this review we will describe the c-MET/HGF signaling pathway in NSCLC, HGF expression as a resistance mechanism to EGFR TKI, and the possible role of HGF inhibition in the treatment of lung cancer patients, focusing specifically on ficlatuzumab.
c-MET/hepatocyte growth factor axis and lung cancer
The c-MET oncogene was first identified in the mid 1980s. It encodes a member of the receptor tyrosine kinase family and is structurally distinct from other components of the family. The receptor is a heterodimer composed of two subunits, the α- and β-chain ().Citation27,Citation28 The α-chain is completely extracellular and is linked to the β-chain by a disulphide bond. The β-chain includes three domains: an extracellular portion, a transmembrane domain, and a cytoplasmic one. The intracellular domain contains a juxtamembrane portion, a tyrosine kinase domain, and a carboxy-terminal tail.Citation27,Citation28
Figure 1 c-MET/HGF pathway.
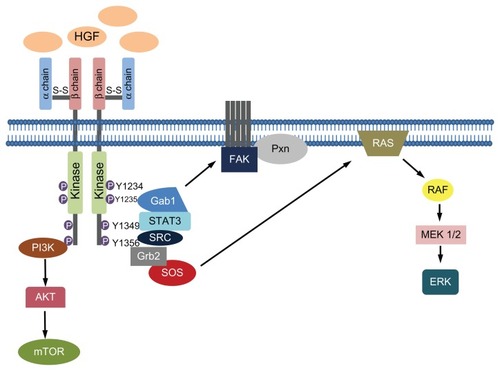
Shortly after the discovery of MET, its physiological ligand, HGF or scatter factor, was identified.Citation29 It is a platelet-derived mitogen for hepatocytes and other normal cell types and a fibroblast-derived factor for epithelial cell scattering, ie, it induces random movement in epithelial cells.Citation29–Citation31 HGF is a morphogen that induces transition of epithelial cells into a mesenchymal morphology. Both tumor and stromal cells have been identified as potential sources of HGF.Citation32 Co-culture studies investigating tumor–stromal interaction demonstrated that fibroblast-dependent carcinoma cell growth and invasion is inhibited by anti-HGF antibodies, highlighting the importance of stroma-derived HGF in tumor sustenance and progression.Citation33 It is synthesized in an inactive form and then converted into a two chain heterodimer, including an amino-terminal domain (N), four Kringle domains (K1–K4), and a serine protease homology domain. The N-K1 portion is responsible for MET binding and dimerization or multimerization. The joining of two or more c-MET receptors leads to phosphorylation of the tyrosine residues Y1234 and Y1235 in the tyrosine kinase domain, and phosphorylation of the residues Y1349 and Y1356 near the carboxy-terminal tail.Citation34 The phosphorylation of the carboxy-terminal tail forms a multifunctional docking site that recruits intracellular adapters and substrates such as STAT3, Grb2, Gab1, PI3K, Shc, Src, Shp2, and Shp1.Citation35 Thus, several pathways involved in proliferation, survival, cell motility, invasion, and metastasis are activated. Interestingly, c-MET activation leads to the recruitment of effectors involved in the epithelial–mesenchymal transition through RAS/MAPK signaling and the FAK/paxillin complex ().
Deregulation of c-MET/HGF signaling may result in carcinogenesis in several solid tumors.Citation36,Citation37 The most common mechanism of activation is c-MET protein expression due to transcriptional upregulation in the absence of gene amplification.Citation38 Receptor overexpression can also be determined by gene amplification.Citation39 Another rare mechanism of activation of the axis is by mutation of the c-MET gene.Citation38 Kinase activation may be ligand independent, but in cancer it is mainly caused by binding of the ligand. Even in the case of c-MET activating mutations, HGF is needed to start the catalytic activity of the receptor.Citation40 HGF plays a fundamental role in the c-MET axis in cancer as it can act either as a paracrine factor, causing positive feedback leading to c-MET transcription,Citation41 or act by an autocrine mechanism.Citation42,Citation43
c-MET gene amplification and overexpression have been associated with poor patient outcome in several studies.Citation44,Citation45 The concentration of HGF in serum or cancer tissue was associated with progression of disease in many cancer types including breast,Citation43,Citation46,Citation47 gastric,Citation48 bladder,Citation49 colorectal,Citation50 SCLC,Citation51 and myeloma.Citation52 Several studies on NSCLC reported intratumoral and plasma HGF levels to be prognostic indicators.Citation53–Citation56 Two research groups analyzed the intratumoral levels of HGF in 56 and 183 resected NSCLC respectively and found an inverse correlation between HGF levels and overall survival, ie, individuals with high levels of intratumoral HGF were likely to have a worse prognosis.Citation53,Citation54 More recently, Hosoda et alCitation55 studied plasma HGF levels in 25 resected NSCLC, revealing a better outcome in terms of both disease free survival (P = 0.032) and overall survival (P = 0.020) for patients with lower levels of HGF. In a similar study by Ujiie et al,Citation56 HGF plasma levels were a negative prognostic factor only for survival (P = 0.016) in a cohort of 109 surgically treated NSCLC patients.
Role of HGF in EGFR TKI resistance and rationale for its blockade
Other than a prognostic indicator, HGF seems to be involved in resistance to agents targeting the EGFR family, not only in lung cancer but also in other malignancies. Recently, our group investigated the role of MET and HGF gene copy number in a large population of metastatic breast cancer patients treated with trastuzumab, an anti-HER2 antibody, and we showed that high MET and HGF gene copy numbers associated with an increased risk of resistance to the anti-HER2 therapy.Citation57 In lung cancer, HGF can independently activate both PI3K/AKT and ERK signaling leading to drug resistance in the presence of EGFR TKI. Unlike MET amplified resistant cancers, HGF-mediated resistance occurs through Gab1 and does not involve HER3.Citation58 In 2008, a Japanese group administered HGF to an adenocarcinoma cell line harboring a sensitizing EGFR gene exon 19 deletion and found that HGF induced resistance in a dose-dependent manner.Citation59 Higher levels of HGF can be detected in tumor specimens from NSCLC patients that are clinically resistant to gefitinib or erlotinib compared to pretreatment tumor specimens. Yano et alCitation60 analyzed HGF expression in paraffin-embedded specimens from 93 EGFR mutant lung cancer patients and found a higher level of HGF expression in tumors with intrinsic and acquired resistance to EGFR TKI. In another study, Turke et alCitation58 compared HGF levels in 16 NSCLC patients for which pre- and post-treatment specimens were available and found that HGF expression was significantly higher in the TKI resistant specimens than in the pretreatment specimens, supporting a role for HGF alone in promoting drug resistance. Both these research groups postulated that HGF may induce EGFR TKI resistance by selection of clones with MET gene amplification.Citation58,Citation60 Recently some researchers investigated whether HGF levels in blood may predict response to EGFR TKI treatment. Several studies analyzed HGF levels in serum from NSCLC patients treated with an EGFR TKI and not selected according to their EGFR mutational status, finding a strong correlation between serum HGF levels and outcome of treatment.Citation61,Citation63
Considering its properties and role as a determinant or promoter of resistance to EGFR TKI, HGF may represent a perfect candidate as a target for treatment. The growth factor is able to induce MET protein overexpression and its blockade may consequently avoid the development of this resistance mechanism in a consistent proportion of patients. Moreover, HGF can convert cancer cells from an epithelial to a mesenchymal phenotype and it is known that lung cancers expressing mesenchymal markers are more resistant to EGFR TKI treatment than tumors with an epithelial phenotype.Citation64,Citation65 A prominent question for all resistance mechanisms is whether they occur as a consequence of treatment or if they exist prior to treatment and are selected under therapy pressure. Growing evidence indicates that resistance mechanisms are already present in small clones of tumor cells, as in the case of secondary EGFR mutationsCitation58 and MET amplification.Citation66 Therefore, treatment with a combination of an EGFR TKI and an anti-HGF or anti-MET agent, particularly in patients with evidence of MET amplification or HGF overexpression, could be more effective than an EGFR TKI alone. In addition, HGF and its receptor are clearly involved in the processes of invasion and metastasis and preclinical data suggest a synergistic effect of EGFR TKI and anti-MET agents, even in EGFR wild-type models.Citation67 For all the listed reasons, anti-MET and anti-HGF strategies are currently under development in NSCLC and other malignancies.
Preclinical data suggested that HGF inhibition could be potentially effective against lung cancer. Okamoto et alCitation68 examined the effects of adding an anti-HGF antibody (TAK-701) to gefitinib treatment in an EGFR mutant cell line engineered to stably express HGF. The combination suppressed cell growth by inhibition of phosphorylation of MET and of the downstream effectors of the EGFR pathway (EGFR, ERK, and AKT), indicating that autocrine c-MET/HGF signaling contributes to gefitinib resistance. In athymic nude mice, the combination therapy of TAK-701 and gefitinib inhibited tumor growth in vivo. In another study, a different anti-HGF antibody (L2G7) was used in combination with gefitinib in HGF expressing mice in which lung tumors were induced by exposure to a carcinogen.Citation69 The mean tumor number in the group treated with the combination of L2G7 and gefitinib was significantly lower than with single agents. Apoptosis was significantly higher in the group treated with combination therapy (17-fold) compared to a single agent (7.9- and 3.5-fold for L2G7 and gefitinib, respectively).
Ficlatuzumab: current status and future directions
Ficlatuzumab (SCH 900105 or AV-299, Aveo Pharmaceuticals, Inc, Cambridge, MA, USA) is a humanized IgG1 antibody that binds the HGF ligand with high affinity and specificity, thus inhibiting c-MET/HGF biological activities. Its pharmacokinetic profile is characterized by a dose-proportional drug exposure with a low systemic clearance and a terminal half-life of 7–10 days.Citation70 In a phase I trial, ficlatuzumab monotherapy resulted in decreases of phospho-MET, phospho-ERK, phospho-AKT, and Ki67.Citation71 The antitumor efficacy of ficlatuzumab was evaluated in paracrine models of a HGF-dependent NSCLC cell line xenografted into SCID mice engineered to produce human HGF. Ficlatuzumab monotherapy decreased tumor growth in a dose-dependent manner and led to significant reductions in phospho-c-MET and phospho-AKT levels, but produced a concurrent increase in phospho-EGFR levels. Therefore, ficlatuzumab was studied in combination with erlotinib or cetuximab, and the two combination treatments showed increased antitumor activity when compared to the single agents.Citation71 The potent antitumor activity of the combination with EGFR inhibitors observed in preclinical models supported further development in NSCLC patients. In a phase I trial, ficlatuzumab was administered both as a single agent (in 24 patients) and in combination with erlotinib 150 mg daily (in 13 patients) in 37 patients with solid tumors, and was well tolerated up to the maximum tested dose of 20 mg/kg every two weeks.Citation72 The most common toxicities of ficlatuzumab monotherapy were fatigue, peripheral edema, headache, and diarrhea; skin rash and diarrhea were the major side effects of the combination treatment. Another phase Ib trial studied ficlatuzumab in association with gefitinib in 15 molecularly unselected Asian NSCLC patients.Citation25 The recommended phase II dose was 20 mg/kg every two weeks for ficlatuzumab and 250 mg daily for gefitinib. Among the twelve patients treated in the 20 mg/kg arm, five were EGFR TKI naïve and all of them reached a partial response. In the same treatment arm, stabilization of disease was observed in four cases and progression in three cases.
Recently, Mok et al presented the results of a randomized phase II trial comparing gefitinib as single agent versus the combination of gefitinib and ficlatuzumab.Citation73 The study enrolled 188 Asian treatment naïve patients with lung adenocarcinoma not selected for EGFR mutational status, even if the study population had clinical characteristics frequently associated with the presence of EGFR mutations. The primary endpoint of the study was to compare the overall response rate between treatment arms. In the overall population there was no statistical difference in response rate (40% for gefitinib arm versus 43% for the combination arm) or progression free survival (4.7 months versus 5.6 months in the gefitinib arm versus combination arm, respectively). Surprisingly, subgroup analyses showed that combination therapy was more effective in patients with low MET expression. In particular, patients with activating mutations in the EGFR gene and low c-MET levels seemed to benefit more from the combination in terms of progression free survival, indicating that c-MET/HGF inhibition may delay the rise of EGFR TKI resistance in this specific population of lung cancer patients. Nevertheless, the low number of patients included in the subgroup analyses precludes any firm conclusion.
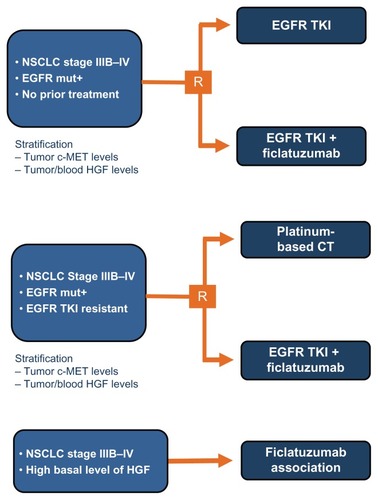
The phase II trial with ficlatuzumab did not reach its primary endpoint but its exploratory biomarker analysis provided important directions for future studies with this agent. In our opinion, anti-MET agents should be explored mainly in three groups of NSCLC patients (): (1) In EGFR TKI naïve patients harboring EGFR mutations in combination with EGFR TKI. Because MET amplification is one of the most relevant mechanisms involved in EGFR TKI resistance,Citation13,Citation74 it is possible that combining an EGFR TKI with an anti-MET agent leads to a delay in tumor progression; (2) In EGFR wild-type patients with high MET expression or MET gene amplification. MET gene amplification is a rare event in NSCLC, occurring in approximately 5% of cases.Citation75 Data with onartuzumab, another anti-MET antibody, suggested that this agent in combination with erlotinib is more effective than erlotinib alone in high MET expressing patients.Citation18 In a phase II randomized trial, tivantinib, a small molecule MET inhibitor, significantly delayed tumor progression when combined with erlotinib in MET amplified patients.Citation23 Overall, data with anti-MET agents indicate that this class of drugs is potentially active in patients selected on the basis of MET expression or gene copy number; and (3) In patients with acquired resistance to EGFR TKI. Preclinical models showed that the combination of an irreversible EGFR TKI with an anti-MET agent is more effective than an anti-MET agent alone in NSCLC cell lines with acquired resistance to reversible EGFR TKI.Citation12 Moreover, we observed that high levels of MET amplification, known to be associated with gefitinib resistance in vitro, rarely occurs in untreated NSCLC, irrespective of EGFR status, and that it may develop only under therapeutic pressure, leading to the conclusion that, in EGFR TKI naïve patients, the level of genomic gain for MET is not increased enough to impact response to TKI. This finding has clinical implications since support for anti-MET strategies should be focused particularly on EGFR TKI resistant patients, where MET gene gain is more frequently observed and can drive tumor resistance.Citation75
Figure 2 Future possible study designs for ficlatuzumab clinical development.
Conclusion
The treatment of advanced NSCLC has deeply changed during the last decade and is rapidly moving toward personalized medicine. Biomarker analysis is becoming more and more important for defining prognosis and for offering the best treatment to our patients. c-MET and HGF have emerged as important biomarkers in NSCLC and other malignancies. Ficlatuzumab is a monoclonal antibody directed against HGF with promising preclinical activity; so far it has not been confirmed in clinical trials. To date, only data from a phase II trial are available and additional studies are planned. Nevertheless, accurate patient selection is of crucial relevance for understanding the real benefit produced by the drug and for offering our patients new therapies positively impacting on survival.
Acknowledgment
In part supported by the Italian Association for Cancer Research (AIRC) and Associazione Oncologia Traslazionale (AOT).
Disclosure
The authors report no conflicts of interest in this work.
References
- JemalABrayFCenterMMWardEFormanDGlobal cancer statisticsCA Cancer J Clin201161699021296855
- LynchTJBellDWSordellaRActivating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinibN Engl J Med20043502129213915118073
- MokTSWuYLThongprasertSGefitinib or carboplatin-paclitaxel in pulmonary adenocarcinomaN Engl J Med200936194795719692680
- LeeJSParkKKimS-WA randomized phase III study of gefitinib (IRESSA) versus standard chemotherapy (gemcitabine plus cisplatin) as a first-line treatment for never-smokers with advanced or metastatic adenocarcinoma of the lungJ Thor Oncol.20094Suppl 19 Abstract PRS 4
- MitsudomiTMoritaSYatabeYGefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trialLancet Oncol20101112112820022809
- MaemondoMInoueAKobayashiKGefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFRN Engl J Med20103622380238820573926
- ZhouCCWuYLChenGErlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 studyLancet Oncol20111273574221783417
- RosellRCarcerenyEGervaisRErlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutations-positive non-small cell lung cancer (EURTAC) a multicenter, open-label, randomized phase 3 trialLancet Oncol20121323924622285168
- YangJC-HSchulerMHYamamotoNLUX-Lung 3: a randomized, open-label, phase III study of afatinib versus pemetrexed and cisplatin as first-line treatment for patients with advanced adenocarcinoma of the lung harboring EGFR-activating mutationsJ Clin Oncol201230Suppl Abstract LBA7500
- KwakELSordellaRBellDWIrreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinibProc Natl Acad Sci U S A20051027665767015897464
- KobayashiSBoggonTJDayaramTEGFR mutation and resistance of non-small-cell lung cancer to gefitinibN Engl J Med200535278679215728811
- BalakMNGongYRielyGJNovel D761Y and common secondary T790M mutations in epidermal growth factor receptor-mutant lung adenocarcinoma with acquired resistance to kinase inhibitorsClin Cancer Res2006126494650117085664
- EngelmanJAZajnullahuKMitsudomiTMET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signalingScience20073161039104317463250
- SequistLVWaltmanBADias-SantagataDGenotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitorsSci Transl Med2011375ra26
- WangWLiQYamadaTCrosstalk to stromal fibroblasts induces resistance of lung cancer to epidermal growth factor receptor tyrosine kinase inhibitorsClin Cancer Res2009156630663819843665
- XuLKikuchiEXuCCombined EGFR/MET or EGFR/HSP90 inhibition is effective in the treatment of lung cancers codriven by mutant EGFR containing T790M and METCancer Res2012723302331122552292
- JanjigianYYSmitEFHornLActivity of afatinib/cetuximab in patients (pts) with EGFR mutant non-small cell lung cancer (NSCLC) and acquired resistance (AR) to EGFR inhibitorsAnn Oncol201223Suppl 9ix401 Abstract 12270
- SpigelDRErvinTJRamlauRFinal efficacy results from OAM4558 g, a randomized phase II study evaluating MetMAb or placebo in combination with erlotinib in advanced NSCLCJ Clin Oncol201129Suppl Abstract 7505
- KwakELCamidgeDRClarkJClinical activity observed in a phase I dose escalation trial of an oral c-met and ALK inhibitor, PF-02341066J Clin Oncol200927Suppl Abstract 3509
- EderJPVande WoudeGFBoernerSALoRussoPMNovel therapeutic inhibitors of the c-Met signaling pathway in cancerClin Cancer Res2009152207221419318488
- YakesFMChenJTanJCabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growthMol Cancer Ther2011102298230821926191
- KollmannsbergerCKHurwitzHVlahovicGPhase I study of daily administration of MGCD265 to patients with advanced malignancies (Study 265-101)J Clin Oncol200927Suppl Abstract e14525
- SequistLVvon PawelJGarmeyEGRandomized phase II study of erlotinib plus tivantinib versus erlotinib plus placebo in previously treated non–small-cell lung cancerJ Clin Oncol2011293307331521768463
- DateKMatsumotoKShimuraHTanakaMNakamuraTHGF/NK4 is a specific antagonist for pleiotrophic actions of hepatocyte growth factorFEBS Lett1997420169450538
- TanEParkKLimWTPhase Ib study of ficlatuzumab (formerly AV-299), an anti-hepatocyte growth factor (HGF) monoclonal antibody (MAb) in combination with gefitinib (G) in Asian patients (pts) with NSCLCJ Clin Oncol201129Suppl 15 Abstract 7571
- MaPCMaulikGChristensenJSalgiaRc-Met: structure, functions and potential for therapeutic inhibitionCancer Metastasis Rev200322209325
- SattlerMMaPCSalgiaRTherapeutic targeting of the receptor tyrosine kinase MetCancer Treat Res200411912113815164876
- NakamuraTNawaKIchiharaAKaiseNNishinoTPurification and subunit structure of hepatocyte growth factor from rat plateletsFEBS Lett19872243113163319692
- GohdaETsubouchiHNakayamaHHuman hepatocyte growth factor in plasma from patients with fulminant hepatic failureExp Cell Res19861661391503527727
- StokerMGherardiEPerrymanMGrayJScatter factor is a fibroblast-derived modulator of epithelial cell mobilityNature19873272392422952888
- JiangWHiscoxSMatsumotoKNakamuraTHepatocyte growth factor/scatter factor, its molecular, cellular and clinical implications in cancerCrit Rev Oncol–Hematol19992920924810226727
- MatsumotoKDateKOhmichiHNakamuraTHepatocyte growth factor in lung morphogenesis and tumor invasion: role as a mediator in epithelium-mesenchyme and tumor-stroma interactionsCancer Chemother Pharmacol199638SupplS42S478765416
- ComoglioPMBoccaccioCScatter factors and invasive growthSeminar Cancer Biol201111153165
- ZhangYWVande WoudeGFHGF/SF-met signaling in the control of branching morphogenesis and invasionJ Cell Biochem20038840841712520544
- NakashiroKHaraSShinoharaYPhenotypic switch from paracrine to autocrine role of hepatocyte growth factor in an androgen-independent human prostatic carcinoma cell line, CWR22RAm J Pathol200416553354015277227
- NaldiniLVignaENarsimhanRPHepatocyte growth factor (HGF) stimulates the tyrosine kinase activity of the receptor encoded by the proto-oncogene c-METOncogene199165015041827664
- MaPCJagadeeswaranRJagadeeshSFunctional expression and mutations of c-Met and its therapeutic inhibition with SU11274 and small interfering RNA in non-small cell lung cancerCancer Res2005651479148815735036
- SmolenGASordellaRMuirBAmplification of MET may identify a subset of cancers with extreme sensitivity to the selective tyrosine kinase inhibitor PHA-665752Proc Natl Acad Sci U S A20061032316232116461907
- MichieliPBasilicoCPennacchiettiSMutant Met-mediated transformation is ligand-dependent and can be inhibited by HGF antagonistsOncogene1999185221523110498872
- BoccaccioCGaudinoGGambarottaGGalimiFComoglioPMHepatocyte growth factor (HGF) receptor expression is inducible and is part of the delayed-early response to HGFJ Biol Chem199426912846128518175699
- TuckABParkMSternsEEBoagAElliottBECoexpression of hepatocyte growth factor and receptor (Met) in human breast carcinomaAm J Pathol19961482252328546209
- KoochekpourSJeffersMRulongSMet and hepatocyte growth factor/scatter factor expression in human gliomasCancer Res199757539153989393765
- NavabRLiuJSeiden-LongICo-overexpression of Met and hepatocyte growth factor promotes systemic metastasis in NCI-H460 non-small cell lung carcinoma cellsNeoplasia2009111292130020019837
- OkudaKSasakiHYukiueHYanoMFujiiYMet gene copy number predicts the prognosis for completely resected non-small cell lung cancerCancer Sci2008992280228519037978
- CappuzzoFMarchettiASkokanMIncreased MET gene copy number negatively affects survival of surgically resected non-small-cell lung cancer patientsJ Clin Oncol2009271667167419255323
- ToiMTaniguchiTUenoTSignificance of circulating hepatocyte growth factor level as a prognostic indicator in primary breast cancerClin Cancer Res199846596649533534
- YamashitaJOgawaMYamashitaSImmunoreactive hepatocyte growth factor is a strong and independent predictor of recurrence and survival in human breast cancerCancer Res199454163016338137271
- TaniguchiTKitamuraMAraiKIncrease in the circulating level of hepatocyte growth factor in gastric cancer patientsBr J Cancer1997756736779043023
- GohjiKNomiMNiitaniYIndependent prognostic value of serum hepatocyte growth factor in bladder cancerJ Clin Oncol2000182963297110944129
- ToiyamaYMikiCInoueYOkugawaYTanakaKKusunokiMSerum hepatocyte growth factor as a prognostic marker for stage II or III colorectal cancer patientsInt J Cancer20091251657166219569242
- BhartiAMaPCMaulikGHaptoglobin alpha-subunit and hepatocyte growth factor can potentially serve as serum tumor biomarkers in small cell lung cancerAnticancer Res2004241031103815154618
- SeidelCBørsetMTuressonIAbildgaardNSundanAWaageAElevated serum concentrations of hepatocyte growth factor in patients with multiple myeloma. The Nordic Myeloma Study GroupBlood1998918068129446640
- SiegfriedJMWeissfeldLASingh-KawPWeyantRJTestaJRLandreneauRJAssociation of immunoreactive hepatocyte growth factor with poor survival in resectable non-small cell lung cancerCancer Res1997574334399012470
- OnitsukaTUramotoHOnoKComprehensive molecular analyses of lung adenocarcinoma with regard to the epidermal growth factor receptor, K-ras, MET and hepatocyte growth factor statusJ Thorac Oncol2010559159620150826
- HosodaHIzumiHTukadaYPlasma hepatocyte growth factor elevation may be associated with early metastatic disease in primary lung cancer patientsAnn Thorac Cardiovasc Surg2012181721959198
- UjiieHTomidaMAkiyamaHSerum hepatocyte growth factor and interleukin-6 are effective prognostic markers for non-small cell lung cancerAnticancer Res2012323251325822843899
- MinutiGCappuzzoFDuchnowskaRIncreased MET and HGF gene copy numbers are associated with trastuzumab failure in HER2-positive metastatic breast cancerBr J Cancer201210779379922850551
- TurkeABZejnullahuKWuYLPreexistence and clonal selection of MET amplification in EGFR mutant NSCLCCancer Cell201017778820129249
- YanoSWangWLiQIHepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor-activating mutationsCancer Res2008689479948719010923
- YanoSYamadaTTakeuchiSHepatocyte growth factor expression in EGFR mutant lung cancer with intrinsic and acquired resistance to tyrosine kinase inhibitors in a Japanese cohortJ Thorac Oncol201162011201722052230
- MasagoKTogashiYFujitaSClinical significance of serum hepatocyte growth factor and epidermal growth factor gene somatic mutations in patients with non-squamous non-small cell lung cancer receiving gefitinib or erlotinibMed Oncol2012291614162121779929
- KasaharaKAraoTSakaiKImpact of serum hepatocyte growth factor on treatment response to epidermal growth factor receptor tyrosine kinase inhibitors in patients with non–small cell lung adenocarcinomaClin Cancer Res2010164616462420679350
- HanJYKimJYLeeSHYooNJChoiBGAssociation between plasma hepatocyte growth factor and gefitinib resistance in patients with advanced non-small cell lung cancerLung Cancer20117429329921440951
- WittaSEGemmillRMHirschFRRestoring E-cadherin expression increases sensitivity to epidermal growth factor receptor inhibitors in lung cancer cell linesCancer Res20066694495016424029
- ThomsonSBuckEPettiFEpithelial to mesenchymal transition is a determinant of sensitivity of non-small-cell lung carcinoma cell lines and xenografts to epidermal growth factor receptor inhibitionCancer Res2005659455946216230409
- FujitaYSudaKKimuraHHighly sensitive detection of EGFR T790M mutation using colony hybridization predicts favorable prognosis of patients with lung cancer harboring activating EGFR mutationJ Thorac Oncol201271640164422899358
- CappuzzoFJannePASkokanMMET increased gene copy number and primary resistance to gefitinib therapy in non-small cell lung cancer patientsAnn Oncol20092029830418836087
- OkamotoWOkamotoITanakaKTAK-701, a humanized monoclonal antibody to hepatocyte growth factor, reverses gefitinib resistance induced by tumor-derived HGF in non-small cell lung cancer with an EGFR mutationMol Cancer Ther201092785279220716641
- StabileLPRothsteinMEKeohavongPTargeting of both the c-Met and EGFR pathways results in additive inhibition of lung tumorigenesis in transgenic miceCancers (Basel)201022153217021390244
- ElezETaberneroJPrudkinLPharmacodynamic (PD) – pharmacokinetic (PK) study of ficlatuzumab (F), a monoclonal antibody (MAB) directed to the hepatocyte growth factor (HGF), in patients (Pts) with advanced solid tumors who have live metastases (Mets)Ann Oncol201223Suppl 9ix154 Abstract 443PD
- MeetzeKABoudrowAConnolyKAnti-tumor activity of SCH 900105 (AV299), an anti-HGF antibody, in non-small cell lung cancer modelsMol Cancer Ther.20098Suppl 12 Abstract C173
- PatnaikAWeissGJPapadopoulosKPhase I study of SCH 900105 (SC), an anti-hepatocyte growth factor (HGF) monoclonal antibody (MAb), as a single agent and in combination with erlotinib (E) in patients (pts) with advanced solid tumorsJ Clin Oncol.201028Suppl Abstract 2525
- MokTSKParkKGeaterSLA randomized phase (Ph) 2 study with exploratory biomarker analysis of ficlatuzumab (F) a humanized hepatocyte growth factor (HGF) inhibitory MAB in combination with gefitinib (G) versus G in Asian patients (pts) with lung adenocarcinoma (LA)Ann Oncol.201223Suppl 9ix391 Abstract 1198P
- EngelmanJAJannePAMechanisms of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancerClin Cancer Res2008142895289918483355
- CappuzzoFJännePASkokanMMET increased gene copy number and primary resistance to gefitinib therapy in non-small-cell lung cancer patientsAnn Oncol20092029830418836087