Abstract
Hereditary angioedema (HAE) resulting from the deficiency of the C1 inhibitor protein is a rare disease, characterized by paroxysms of edema formation in the subcutis and in the submucosa. Edema can cause obstruction of the upper airway, which may lead to suffocation. Prompt elimination of edema is necessary to save patients from this life-threatening condition. Essentially, these edematous attacks are related to the activation of the kinin-kallikrein system and the consequent release of bradykinin. Ecallantide (known as DX-88 previously), a potent and specific inhibitor of plasma kallikrein is an innovative medicinal product. This is the only agent approved recently by the FDA for all localizations of edematous HAE attacks. Its advantages include no risk of viral contamination, high selectivity, very rapid onset of action, good tolerability, and straightforward subcutaneous administration. Owing to the risk of anaphylaxis, ecallantide should be administered by a health care professional. A postmarketing survey to improve risk-assessment and risk-minimization has been launched. The results of these studies may lead to the approval of ecallantide for self-administration.
Introduction to hereditary angioedema – epidemiology and management
Angioedema
The term ‘angioedema’ means recurrent episodes of sudden, occasionally painful swelling of the subcutis and deeper tissues without accompanying erythema and itching; it may also evolve in the submucosa of the upper airways or the gastrointestinal tract. Its pathomechanism involves the role of vasoactive mediators enhancing the permeability of subcutaneous/submucosal capillaries and postcapillary venules – the resulting outflow of plasma into the interstitium produces local edema.Citation1 These mediators increasing vascular permeability may be released from mastocytes or basophilic cells (through IgE-mediated processes or direct cell activation), through activation of the kinin system, or through mechanisms not yet clear.Citation2
Hereditary angioedema
Classification and epidemiology
This inherited form of bradykinin-mediated angioedema is an autosomal dominant disorder with three distinct types. Hereditary angioedema (HAE-C1-INH) resulting from deficiency of the C1 inhibitor is caused by a mutation in the C1 INH gene and is manifested as two phenotypes.Citation3 More than 200 different mutations of the C1INH gene have been described to date.Citation4
In type I HAE-C1-INH, the defective gene does not produce a protein. Type II is characterized by the production of a dysfunctional inhibitor – therefore, the concentration of C1-INH protein may be normal or often elevated, but this protein lacks function. Type I is more prevalent, occurring in approx. 85% of cases and type II is seen in the remaining 15 per cent. The estimated prevalence of HAE-C1-INH is 1:10,000 to 1:50,000 according to international data.Citation5 The serine protease C1-INH is involved in the regulation of the complement, coagulation, fibrinolytic, and kinin plasma cascade systems.Citation6 These systems undergo activation in C1-INH deficiency and the vasoactive mediator bradykinin (produced from high-molecular-weight kininogen during activation of the kallikrein-kinin system) is responsible for the extravasation of plasma and the occurrence of characteristic disease symptoms.Citation7–Citation9 A third type of hereditary angioedema has been described recently. In this type (HAEIII-FXII), plasma level and functional activity of the C1-INH protein are both normal and although missense mutation of the factor XII gene can be demonstrated in approximately 30% of cases, the mechanism of edema formation is unknown. The clinical manifestations of type III HAE are identical to those seen in HAE caused by C1-INH deficiency.Citation10 The following discussion and findings all pertain to HAE-C1-INH.
Diagnosis
The diagnosis is established through evaluation of family history and clinical manifestations, complement testing, and genetics analysis.
Family history
In about 75% of HAE cases, carriers of a mutated C1INH gene are found among first-degree relatives. Family members of the remaining 25% of patients are not afflicted by this abnormality and therefore, their disease is probably caused by de novo mutations.Citation5
Clinical symptoms
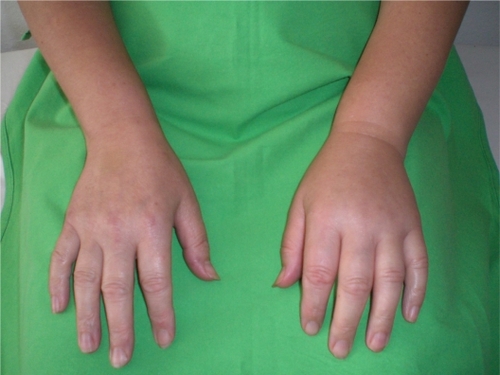
Time of onset, incidence, location and severity of clinical manifestations all exhibit inter-individual variation. Attacks begin to occur at around entering school age. Half of the afflicted patients experience the initial attack during the first 10 years, whereas an additional third of patients during the second decade of life.Citation11 Certain factors with a possible role in inducing attacks can be identified in a proportion of patients and edematous episodes (including mechanical trauma, menstruation, pregnancy, infections, oral contraceptives, angiotensin-converting enzyme inhibitors).Citation12 Edema can develop on the extremities, trunk, genitals, and face, but also in the mucosa of the upper airways or of the intestines. Cutaneous edema may be accompanied by pain and deformity (). Facial edema may cause severe disfiguration with consequent limitation of routine activities, whereas loss of function from edema of the extremities may interfere with work and activities of daily living. The symptoms of abdominal attacks include colicky pain, nausea, vomiting, and profuse, watery diarrhoea occurring after the resolution of the attack. These symptoms can mimic the clinical manifestations of the ‘acute abdomen’ and therefore, patients with these manifestations frequently undergo unnecessary surgery during their abdominal attack.Citation13,Citation14 Upper airway edema (UAE) may lead to asphyxia by causing airway obstruction with 30% to 50% mortality in undiagnosed or inappropriately managed cases, especially those following a fulminant course.Citation15,Citation16
Figure 1 Subcutaneous edema on the left hand of a patient with HAE-C1-INH.
Complement and genetic studies
HAE-C1-INH can be diagnosed with complement testing; molecular genetics analysis is helpful in uncertain cases; it is an important tool in prenatal diagnostics. Type I HAE-C1-INH is characterized by lower C4, as well as reduced antigenic and functional C1-INH levels. In Type II HAE-C1-INH, C4 is reduced, and antigenic C1-INH level is high or normal, and the functional activity of C1-INH is impaired.Citation17,Citation18 Early diagnosis and timely introduction of follow-up care are indispensable for preserving the patients’ quality of life.
Management
As HAE is a hereditary disorder, gene therapy would be the optimal and definite solution; however, it is not yet available. The guidelines on the management of HAE have been developed through international consensus.Citation17,Citation19 The initial step of the management of HAE-C1-INH is counseling and education, as these enable the patient to avoid potential triggering factors of attacks as much as possible. If, nevertheless, edematous attacks recur frequently, prophylaxis should be introduced.Citation20
Long-term prophylaxis is implemented by administering antifibrinolytics (tranexamic acid, epsilon-aminocaproic acid) or attenuated androgens (stanozolol, danazol, oxan-drolone, methyltestosterone), although their mode of action is still not precisely known.Citation21,Citation22 The use of these drugs – and of attenuated androgens, in particular – is limited by the risk of side effects, like virilization, liver enzyme elevation, liver tumors and dyslipidemia.Citation23 A proportion of the latter can be avoided at least partially by administering the lowest effective dose, and by their timely recognition as afforded by periodic laboratory screens (complete blood count, liver enzyme activity, creatine kinase, urinalysis) and abdominal ultrasound imaging. Drug adverse reactions lead to discontinuation of treatment in certain cases.Citation24,Citation25 The agents appropriate for long-term prophylaxis also include human plasma-derived C1-INH concentrate.Citation26,Citation27 A multicenter clinical study was conducted only with the nanofiltrated product (Cinryze®, Viropharma), which has been approved by the FDA for the prevention of edematous attacks in adolescents and adults. This drug has been available in the US since 2009.Citation28
Short-term prophylaxis is recommended for patients undergoing surgical or diagnostic interventions on the head or neck.Citation29 In this case, attenuated androgens or antifibrinolytic agents should be administered in higher doses before the intervention, as well as for 4 to 5 days thereafter. However, the most effective prophylaxis is C1-INH replacement with the concentrate administered 1 hour (less than 6 hours) before the intervention.Citation17
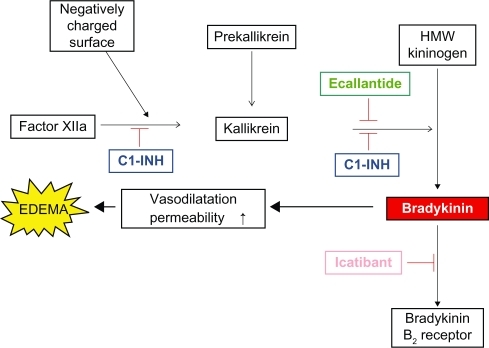
Nowadays, there are several options available for the acute treatment of edematous attacks ().Citation30 The largest body of experience exists with human plasma-derived C1-INH concentrate.Citation31 Two intravenous products are currently marketed. One of them is Berinert P® (Berinert® in the US) produced by CSL Behring is available in 28 countries worldwide.Citation32 In the US, it was approved by the FDA in 2009 for the treatment of facial and abdominal edema in adolescents and adults.Citation33 A prospective study has proven the efficacy and safety of C1-INH concentrate in laryngeal HAE attacks.Citation34 The other one, Cetor®, manufactured by the Dutch not-for-profit Sanquin Blood Supply Foundation, is another blood-derived, purified, nanofiltrated C1-INH concentrate that has been used for the emergency therapy of HAE since more than 30 years. Recombinant C1-INH (obtained from the milk of transgenic rabbits) for intravenously use (developed by Pharming Group NV), was approved by the EMEA in 2010 for treatment of all types of HAE attacks in adults. It has been launched in some countries by Swedish Orphan Biovitrum (SOBI) under the brand name Ruconest®.Citation35,Citation36 Icatibant, a bradykinin receptor antagonist (a synthetic decapeptide, which is a selective and specific antagonist of bradykinin B2 receptors) intended for subcutaneous administration is available in several European countries. Although the evaluation of this drug has been initiated by Jerini AG, its clinical development was completed, and the product is being marketed currently by Shire under the brand name Firazyr®. It has been approved by the EMEA for the management of acute edematous attacks (regardless of localization) in adults; Firazyr® is not approved in the US.Citation37
Figure 2 Modes of action of agents appropriate for controlling edematous attacks.
In 2011, the European Commission approved Firazyr® for self-administration by patients who have mastered the technique of subcutaneous injection under the supervision of a health care professional. Of all the products licensed in Europe for the treatment of acute episodes of type I and type II HAE, Firazyr® is the first and only treatment licensed for self-administration.
Ecallantide (Kalbitor®, known as DX-88 previously) is a recombinant kallikrein inhibitor developed by Dyax Corporation.Citation38–Citation41 This is the only product approved recently by the FDA for all localizations of edematous HAE attacks occurring in patients over 16 years of age. This drug has been available in clinical practice since 2009 in the US.Citation42 When none of these state-of-the-art treatment modalities is available, fresh frozen plasma may be administered if clearly indicated. However, it should be taken into account that the use of solvent/detergent fresh frozen plasma (SDP; preferred over FP/FFP to avoid viral transmission) could theoretically worsen attacks. This is because it contains proteins that mediate edema formation; moreover, its administration remains controversial as there are no clinical trials to support its use.Citation17,Citation43
The following is a summary of results from clinical studies evaluating the pharmacology, mode of action, and pharmacokinetics of ecallantide.
Introducing ecallantide
Ecallantide (company code DX-88) is a potent, specific and reversible plasma kallikrein inhibitor (Ki = 25 pM). Ecallantide was generated via phage display – a technique for the rapid identification of target-specific protein binders – and a library consisting of rationally designed variants of the first Kunitz domain of human tissue factor pathway inhibitor. Ecallantide is a small, 60 amino-acid, 7062 Da protein, produced by recombinant technology (expression in the yeast Pichia pastoris) and purified by chromatography.Citation44
Action
Angioedema attacks caused by C1-INH deficiency are mediated by the excessive release of vasoactive bradykinin. This release is initiated by the uncontrolled activation of factor XII, a protein that autoactivates upon exposure to negatively charged surfaces. The activation of factor XII yields factor XIIa and kallikrein. C1-INH inhibits both proteases. Kallikrein can reciprocally activate more FXII and liberate bradykinin. The mechanistic trigger for the initial activation of plasma kallikrein in vivo is unknown at present, but the result is the cleavage of high-molecular-weight kininogen (HMWK) by plasma kallikrein with the release of bradykinin. Bradykinin acts on the vasculature to increase capillary and endothelial permeability, resulting in extravasation of fluid, which produces the characteristic signs and symptoms of HAE attacks. Acting upon upstream elements in the kinin pathway, ecallantide can produce rapid, specific, complete, and reversible blockade of plasma kallikrein, thereby reducing the excess of endogenous bradykinin.
Chemistry, dosage and administration
Ecallantide is a clear, sterile solution. Each vial contains 10 mg ecallantide as the active substance along with nonpyrogenic inactive ingredients. Ecallantide is preservative-free, with a pH of approximately 7.0; it is stored under refrigeration and should be protected from light. Its recommended dose is 30 mg (3.0 mL) administered subcutaneously in divided, 10 mg doses; injection sites should be distant from the location of angioedema. A 30 mg dose is supplied as three vials, each containing 1 mL of 10 mg/mL ecallantide. If an attack persists, an additional dose of 30 mg may be administered within 24 hours. Ecallantide should only be administered by a health care professional with appropriate medical resources to manage anaphylaxis and hereditary angioedema.Citation45
Pharmacokinetics and metabolism
In the initial Phase I study, subjects received dose-ranging intravenous (IV) infusions. Peak concentrations occurred 2 to 3 hours after subcutaneous (SC) administration, although absorption was slightly slower in heavier patients. Following the administration of a single 30 mg SC dose of ecallantide to healthy subjects, a mean (± standard deviation) maximum plasma concentration of 586 ± 106 ng/mL was observed approximately 2 to 3 hours postdose. The mean area under the concentration-time curve was 3017 ± 402 ng h/mL. Following administration, plasma concentration declined with a mean elimination half-life of 2.0 ± 0.5 hours. Plasma clearance was 153 ± 20 mL/minute and the volume of distribution was 26.4 ± 7.8 L. In patients and in healthy subjects, ecallantide was shown to distribute rapidly in the vascular compartment following either IV or SC administration. This rapid distribution was consistent with the goal of plasma kallikrein inhibition. The population pharmacokinetics of ecallantide has been investigated in 35 patients with HAE (24 females and 11 males) and in 62 healthy subjects, (34 females and 28 males) aged 11 to 68 years. No difference in pharmacokinetics was noted between these two groups. The clearance of ecallantide was 7.56 L/hour, with a steady-state volume of distribution of 15.1 L. Inter-individual variability was 38% for clearance. Ecallantide was cleared rapidly, with an effective half-life of 0.8 to 4.5 hours. Given the short half-life, once-daily administration of multiple doses did not result in accumulation, and repeated-dose pharmacokinetics was similar to that seen after single doses. Age, sex, and baseline creatinine clearance of patients had no impact on ecallantide pharmacokinetics. No pharmacokinetic data are available in patients or individuals with hepatic or renal impairment.Citation44
Pharmacodynamics
No exposure-response relationships have been established between ecallantide and the components of complement or kallikrein-kinin pathways. The effect of ecallantide on activated partial thromboplastin time (aPTT) was measured in view of a potential effect on the intrinsic coagulation pathway. Prolongation of aPTT was observed following intravenous dosing with KALBITOR® (ecallantide) at doses of approximately 20 mg/m2. When 80 mg was administered intravenously to healthy subjects, aPTT increased approximately two-fold over baseline values and returned to normal by 4 hours postdose. No significant QT prolongation has been observed in patients taking ecallantide. In a randomized, placebo-controlled trial (EDEMA4) studying the 30 mg subcutaneous dose vs placebo, 12-lead ECG tracings were obtained at baseline, as well as 2 hours and 4 hours postdose (covering the period to the expected Cmax), and at follow-up (day 7). ECGs were evaluated for PR interval, QRS duration, and QTc interval. Ecallantide had no significant effect on QTc interval, heart rate, or any other components of the ECG (DX-88 [ecallantide]).
Clinical studies on the efficacy and safety of ecallantide
Efficacy
The results of the first placebo-controlled human study of a therapeutic intervention to mitigate the symptoms of HAE attacks was published in 2007.Citation40 This double-blind, placebo-controlled, ascending-dose study assessed the efficacy and tolerability of ecallantide administered intravenously in different doses (5, 10, 20, or 40 mg/m2) to individuals experiencing HAE attacks (N = 49). Ecallantide ameliorated the symptoms of HAE attacks in 72.5% (29/40) of the patients treated, whereas 25.0% (2/8) of placebo patients reported significant improvement of symptoms within 4 hours (P = 0.0169). Ecallantide was well-tolerated at all doses.
The safety and efficacy of ecallantide were evaluated in two randomized, double-blind, placebo-controlled trials (EDEMA3 and EDEMA4) in 168 patients with HAE-C1-INH.
In both trials, the effects of ecallantide were appraised using the Mean Symptom Complex Severity (MSCS) score and the Treatment Outcome Score (TOS). The MSCS score is based on patient-assessed symptom severity (on a scale ranging from 0 – normal to 3 – severe) measured at baseline, 4 hours, and 24 hours. A decrease in MSCS score reflects an improvement in symptoms. The TOS is based on patient assessment of response characterized by the change in symptom severity vs baseline (on a scale ranging from 100 – significant improvement to −100 – significant worsening) measured at 4 hours and 24 hours. A TOS value >0 reflects an improvement in symptoms from baseline.
The EDEMA3 trial was a multicenter, randomized, double-blind, placebo-controlled Phase III study. 160 patients were enrolled and 72 patients (aged 10 years or older) who had experienced an attack of HAE at any anatomical location with at least one moderate or severe symptom were randomized. In a 1:1 ratio, they received either subcutaneous ecallantide at a dose of 30 mg, or placebo (phosphate-buffered saline, pH 7.0) for attacks of HAE (36 in the ecallantide group and 36 in the placebo group). An open-label dose of 30 mg of subcutaneous ecallantide was allowed for cases with severe upper-airway obstruction. Patients were observed for at least 4 hours after administration of the study drug. Final assessment of symptoms was done at 24 hours. The primary endpoint was TOS at 4 hours and the key secondary efficacy endpoint was the change from baseline in MSCS score at 4 hours.
At 4 hours after dosing with the study drug, median TOS was 50.0 for the ecallantide-treated group and 0.0 for placebo-treated patients. At 4 hours after administration of the study-drug, median change in the MSCS score was −1.00 in the ecallantide group and −0.50 in the placebo group. Median time to significant improvement in overall response was 165.0 minutes with ecallantide and >240 minutes with placebo.
At 24 hours after administration of the study-drug, median TOS was 75.0 for the ecallantide group and 0.0 for the placebo group. Median change in the MSCS score at 24 hours was −1.00 with ecallantide and −0.50 with placebo. Median time to sustained improvement in overall response was 67.0 minutes in the ecallantide group and 165.0 minutes in the placebo group.
Compared to placebo, ecallantide significantly reduced the outcome score at 4 hours (the primary endpoint), and the mean symptom complex severity score at 4 hours, as well as both of these at 24 hours. In the placebo group, 36%, whereas in the ecallantide group 14% of patients required medical intervention within 24 hours to treat persisting symptoms.Citation46 The design of the EDEMA4 trial (double-blind, placebo-controlled Phase III study evaluating the treatment of patients with a moderate-to-severe HAE attack) was similar to that of EDEMA3. Patients were randomized 1:1 to receive 30 mg of subcutaneous ecallantide or placebo. Ninety-six patients were enrolled. TOS 4 hours after dosing was 53.4 with ecallantide vs 8.1 with placebo. Mean (SD) change from baseline in MSCS 4 hours after dosing was significantly greater with ecallantide (−0.8), than with placebo (−0.4). At 24 hours, patients treated with ecallantide exhibited a greater decrease from baseline in the MSCS score than with placebo (−1.5 vs −1.1) and a greater TOS, than with placebo (89 vs 55). More patients in the placebo group than in the group receiving ecallantide required medical intervention to treat unresolved symptoms within 24 hours (50% vs 33%).
In the EDEMA4 trial, patients treated with a single subcutaneous dose of ecallantide showed a greater decrease of MSCS vs baseline and a greater TOS than that seen in placebo-treated patients; the differences were statistically significant.Citation47,Citation48 Riedl et al evaluated the promptness of the effect of ecallantide vs placebo using pooled data from 143 participants (70 receiving ecallantide and 73 receiving placebo for 64 abdominal, 55 peripheral and 24 laryngeal attacks) from two randomized, double-blind, placebo-controlled Phase III trials. The quickest onset of the therapeutic effect of ecallantide was demonstrated for abdominal attacks, followed by laryngeal and peripheral attacks.Citation49
Summary of efficacy: Ecallantide was found to be superior to placebo in relieving symptoms, decreasing the severity of attacks, and shortening their duration.
Safety and tolerability
Subcutaneous ecallantide 30 mg was generally well-tolerated by patients with attacks of HAE during the EDEMA3 and EDEMA4 trials. Most adverse events were mild to moderate in severity, and no event that was more common in ecallantide than in placebo recipients occurred in >10% of patients.Citation48 Overall, treatment-emergent adverse events (TEAEs) occurred at a low rate and were manageable. The most common adverse events (≥5%) were headache, nausea, fatigue, diarrhea, upper respiratory tract infection, HAE, nasopharyngitis, vomiting, upper abdominal pain, and pyrexia; most were mild or moderate in severity. Transient, mild, local injection-site reactions were reported in a few patients only. The most clinically relevant TEAE was hypersensitivity to ecallantide. Overall, 13 patients experienced symptoms of possible hypersensitivity during the clinical studies. Four of these patients exhibited symptoms suggestive of anaphylactoid or anaphylactic reactions, such as pruritus, urticaria, allergic rhinitis, throat irritation, pharyngeal edema, flushing, wheezing, rhinorrhea, and occasionally hypotension, within 15 minutes after dosing. These symptoms resolved either spontaneously or following treatment (eg, with antihistamines, epinephrine) in all patients.
Repeated doses of ecallantide were well-tolerated. Of the 219 patients in the program, 19 (8.7%) received five to nine doses and an additional 12 patients (5.5%) received more than nine doses of ecallantide.
Patients developed antibodies to ecallantide. Anti-ecallantide antibodies – of all classes and of IgE type against host cell proteins (Pichia pastoris) – did not appear to correlate with the occurrence of adverse events. Overall, 7.4% of patients seroconverted to anti-ecallantide antibodies. The incidence of seroconversion seemed to increase with increasing drug exposure.Citation46,Citation47
No information is available on drug interactions, as no relevant studies have been conducted. Similarly, no clinical data exist on the management of pregnant women with HAE. The safety and effectiveness of ecallantide in patients under 16 years of age have not been established. It is not known whether ecallantide is excreted in human breast milk. There are no animal or human studies to assess the carcinogenic or mutagenic potential of ecallantide.
Patients’ perspectives: quality of life, patient satisfaction/acceptability, adherence and uptake
The management of HAE patients has not changed over decades. Appropriate therapies for controlling edematous attacks were nonexistent in many countries, including the US. In recent years, however, a variety of innovative medicinal products have been developed, and their efficacy and safety were demonstrated in double-blind, placebo-controlled studies. Simultaneously, these treatment modalities have become available for clinical use in an increasing number of countries and hence, for an increasing number of patients. This is of extreme importance, as the mortality of HAE is high even today, owing to delayed diagnosis and lack of appropriate therapy, primarily. The range of available therapies expanded on one hand and became extremely variegated on the other. Novel agents differ from each other in their manufacturing technology, mode of action, and dosage – this affords to find the best treatment modality for every patient. Individualized patient management is the key to developing an optimal and cost-effective therapeutic strategy.Citation50
Helping patients to feel secure is essential. Prerequisites to achieving this include awareness of this disorder, making an early diagnosis, choosing appropriate treatment, informing patients on the nature of HAE, as well as providing regular follow-up care. The feeling of safety is further enhanced by having the medication at hand, ready for the emergency treatment of edematous attacks anytime and anywhere – this is important, as the onset of edema cannot be predicted in advance. Long-term prophylaxis cannot eliminate all future attacks, including life-threatening edema involving the upper airways. Additional problems include undesirable effects and insufficient efficacy of prophylactic agents. Prompt management of abdominal attacks can help avoiding unnecessary surgery. The optimized management of HAE may reduce the cost of care, improve the patients’ quality of life, cut absenteeism from school and work, and ease the limitations in activities of daily living. Ecallantide is a new, effective agent in this range of therapies.Citation42 The advantages of ecallantide are that it poses no risk of viral contamination; it is highly selective and has a quick onset of action. It is also well-tolerated by patients and can be administered subcutaneously. Its administration is simple, straightforward, and prompt – all these properties are important as timely intervention can prevent the escalation of the attack. Rapid resolution of edema enables patients to resume everyday activities and work as soon as possible. In clinical practice, the administration of ecallantide is recommended only under the guidance and supervision of a health care professional, in facilities where appropriate preconditions exist to recognize and manage any potential anaphylactic reaction. Dyax proposes implementing a postmarketing risk management program (Risk Evaluation and Management Strategy) ensuring the safe use of ecallantide to assess frequency and possible risk.Citation51 It is hoped that these studies will yield positive results and lead to the approval of ecallantide for self-administration by patients – this would result in a substantial improvement in their quality of life. If ecallantide could be administered anytime and anywhere, repeatedly and immediately – ie, without delays caused by finding an appropriate health care professional and institution – it would significantly improve the patients’ quality of life while being cost-effective.
Disclosure
The authors report no conflicts of interest in this work.
References
- KaplanAPGreavesMWAngioedemaJ Am Acad Dermatol2005533373388 quiz38939216112343
- BasMAdamsVSuvoravaTNiehuesTHoffmannTKKojdaGNonallergic angioedema: role of bradykininAllergy200762884285617620062
- DonaldsonVHEvansRRA biochemical abnormality in hereditary angioneurotic edema: absence of serum inhibitor of C’ 1-esteraseAm J Med196335374414046003
- HAEdb, C1 inhibitor gene mutation database Home page on the Internet. [Updated 2008 July 18]. Available at: http://hae.enzim.hu.
- AgostoniACicardiMHereditary and acquired C1-inhibitor deficiency: biological and clinical characteristics in 235 patientsMedicine (Baltimore)19927142062151518394
- DavisAE3rdHereditary angioedema: a current state-of-the-art review, III: mechanisms of hereditary angioedemaAnn Allergy Asthma Immunol20081001 Suppl 2S7S1218220146
- FieldsTGhebrehiwetBKaplanAPKinin formation in hereditary angioedema plasma: evidence against kinin derivation from C2 and in support of “spontaneous” formation of bradykininJ Allergy Clin Immunol198372154606222104
- NussbergerJCugnoMAmstutzCCicardiMPellacaniAAgostoniAPlasma bradykinin in angio-oedemaLancet19983519117169316979734886
- KaplanAPGhebrehiwetBThe plasma bradykinin-forming pathways and its interrelationships with complementMol Immunol201047132161216920580091
- BorkKWulffKHardtJWitzkeGStaubachPHereditary angioedema caused by missense mutations in the factor XII gene: clinical features, trigger factors, and therapyJ Allergy Clin Immunol2009124112913419477491
- BorkKMengGStaubachPHardtJHereditary angioedema: new findings concerning symptoms, affected organs, and courseAm J Med2006119326727416490473
- KempJGCraigTJVariability of prodromal signs and symptoms associated with hereditary angioedema attacks: a literature reviewAllergy Asthma Proc200930549349919843403
- BorkKStaubachPEckardtAJHardtJSymptoms, course, and complications of abdominal attacks in hereditary angioedema due to C1 inhibitor deficiencyAm J Gastroenterol2006101361962716464219
- FarkasHHarmatGKaposiPNUltrasonography in the diagnosis and monitoring of ascites in acute abdominal attacks of hereditary angioneurotic oedemaEur J Gastroenterol Hepatol200113101225123011711780
- BorkKResselNSudden upper airway obstruction in patients with hereditary angioedemaTransfus Apher Sci200329323523814572815
- FarkasHManagement of upper airway edema caused by hereditary angioedemaAllergy Asthma Clin Immunol2010611920667122
- BowenTCicardiMFarkasHInternational consensus algorithm for the diagnosis, therapy and management of hereditary angioedemaAllergy Asthma Clin Immunol2010612420667127
- GompelsMMLockRJAbinunMC1 inhibitor deficiency: consensus documentClin Exp Immunol2005139337939415730382
- LonghurstHJFarkasHCraigTHAE international home therapy consensus documentAllergy Asthma Clin Immunol2010612220667125
- CraigTRiedlMDykewiczMSWhen is prophylaxis for hereditary angioedema necessary?Ann Allergy Asthma Immunol2009102536637219492656
- FrankMMUpdate on preventive therapy (prophylaxis) of hereditary angioedemaAllergy Asthma Proc2011321172121262094
- ZurawBLHereditary angiodema: a current state-of-the-art review, IV: short- and long-term treatment of hereditary angioedema: out with the old and in with the new?Ann Allergy Asthma Immunol20081001 Suppl 2S13S1818220147
- SzeplakiGVargaLValentinSAdverse effects of danazol prophylaxis on the lipid profiles of patients with hereditary angioedemaJ Allergy Clin Immunol2005115486486915806011
- BorkKBygumAHardtJBenefits and risks of danazol in hereditary angioedema: a long-term survey of 118 patientsAnn Allergy Asthma Immunol2008100215316118320917
- FarkasHCzallerICsukaDThe effect of long-term danazol prophylaxis on liver function in hereditary angioedema-a longitudinal studyEur J Clin Pharmacol201066441942620024535
- BorkKWitzkeGLong-term prophylaxis with C1-inhibitor (C1 INH) concentrate in patients with recurrent angioedema caused by hereditary and acquired C1-inhibitor deficiencyJ Allergy Clin Immunol19898336776822926086
- KreuzWMartinez-SaguerIAygoren-PursunERusickeEHellerCKlingebielTC1-inhibitor concentrate for individual replacement therapy in patients with severe hereditary angioedema refractory to danazol prophylaxisTransfusion20094991987199519497056
- ZurawBLBussePJWhiteMNanofiltered C1 inhibitor concentrate for treatment of hereditary angioedemaN Engl J Med2010363651352220818886
- SzemaAMPazGMerriamLStellaccioFJenJModern preoperative and intraoperative management of hereditary angioedemaAllergy Asthma Proc200930333834219368763
- ZurawBLHAE therapies: past present and futureAllergy Asthma Clin Immunol2010612320667126
- FarkasHJakabLTemesszentandrasiGHereditary angioedema: a decade of human C1-inhibitor concentrate therapyJ Allergy Clin Immunol2007120494194717761272
- CSL Behring Global Home page on the Internet. [Updated 2011]. Available at: http://www.cslbehring.com/PRelease/Berinert-Approved.htm.
- CraigTJLevyRJWassermanRLEfficacy of human C1 esterase inhibitor concentrate compared with placebo in acute hereditary angioedema attacksJ Allergy Clin Immunol2009124480180819767078
- CraigTJWassermanRLLevyRJProspective study of rapid relief provided by C1 esterase inhibitor in emergency treatment of acute laryngeal attacks in hereditary angioedemaJ Clin Immunol201030682382920635155
- VargaLFarkasHrhC1INH: a new drug for the treatment of attacks in hereditary angioedema caused by C1-inhibitor deficiencyExpert Rev Clin Immunol20117214315321426252
- ZurawBCicardiMLevyRJRecombinant human C1-inhibitor for the treatment of acute angioedema attacks in patients with hereditary angioedemaJ Allergy Clin Immunol20101264821827 e81420920772
- CicardiMBanerjiABrachoFIcatibant, a new bradykinin-receptor antagonist, in hereditary angioedemaN Engl J Med2010363653254120818888
- WilliamsABairdLGDX-88 and HAE: a developmental perspectiveTransfus Apher Sci200329325525814572818
- LevyJHO’DonnellPSThe therapeutic potential of a kallikrein inhibitor for treating hereditary angioedemaExpert Opin Investig Drugs200615910771090
- SchneiderLLumryWVeghAWilliamsAHSchmalbachTCritical role of kallikrein in hereditary angioedema pathogenesis: a clinical trial of ecallantide, a novel kallikrein inhibitorJ Allergy Clin Immunol2007120241642217559913
- LehmannAEcallantide (Dyax/Genzyme)Curr Opin Investig Drugs200673282290
- BernsteinJAQaziMEcallantide: its pharmacology, pharmacokinetics, clinical efficacy and tolerabilityExpert Rev Clin Immunol201061293920383888
- PremattaMGibbsJGPrattELStoughtonTRCraigTJFresh frozen plasma for the treatment of hereditary angioedemaAnn Allergy Asthma Immunol200798438338817458436
- Advisory Committee Briefing Document, Kalbitor® (ecallantide) For Acute Attacks of Hereditary Angioedema (BLA 125277)0212009 Available at: http://www.fda.gov/downloads/advisorycommit-tees/committeesmeetingmaterials/drugs/pulmonary-allergydrugsadvi-sorycommittee/ucm170334.pdf.
- DX-88 (ecallantide) 1.4.1. Draft Label- KALBITOR®27112009 Available at: http://mms.businesswire.com/bwapps/mediaserver/ViewMedia?mgid=206775&vid=1.
- CicardiMLevyRJMcNeilDLEcallantide for the treatment of acute attacks in hereditary angioedemaN Engl J Med2010363652353120818887
- LevyRJLumryWRMcNeilDLEDEMA4: a phase 3, double-blind study of subcutaneous ecallantide treatment for acute attacks of hereditary angioedemaAnn Allergy Asthma Immunol2010104652352920568386
- Garnock-JonesKPEcallantide: in acute hereditary angioedemaDrugs201070111423143120614949
- RiedlMCampionMHornPTPullmanWEResponse time for ecallantide treatment of acute hereditary angioedema attacksAnn Allergy Asthma Immunol20101056430436 e221130380
- WilsonDABorkKSheaEPRentzAMBlausteinMBPullmanWEEconomic costs associated with acute attacks and long-term management of hereditary angioedemaAnn Allergy Asthma Immunol2010104431432020408341
- StolzLEHornPTEcallantide: a plasma kallikrein inhibitor for the treatment of acute attacks of hereditary angioedemaDrugs Today (Barc)201046854755520830315