
Abstract
Degos disease is characterized by atrophic porcelain-white papules with peripheral erythema. Degos-like lesions have been reported in association with several connective tissue diseases (CTDs), mostly systemic lupus erythematosus (SLE), and rarely in dermatomyositis and systemic sclerosis. Herein, we report three cases of Degos-like lesions in CTDs. Two cases had Degos-like lesions linked to dermatomyositis. Both cases presented with severe inflammatory myopathy without pathognomonic cutaneous signs for dermatomyositis and lacked myositis-specific autoantibodies. Our third case, to our knowledge, is the first case of Degos-like lesions in association with overlapping systemic sclerosis and SLE without major organ involvement. The clinical presentation and histopathology findings of our cases support that Degos-like lesions may not be a specific entity but can be considered as a unique clinical pattern expressed in patients with CTDs.
Introduction
Degos disease or malignant atrophic papulosis is a syndrome of vascular occlusive disorder of unknown cause that mainly involves the skin, gastrointestinal tract, and central nervous system.Citation1,Citation2 Cutaneous finding in Degos disease is characterized by the presence of pathognomonic skin lesions, commonly described as atrophic porcelain-white center papules with rim of peripheral erythema and telangiectasia.Citation3
Degos-like lesions have been reported in association with several CTDs, mainly systemic lupus erythematosus (SLE). To date, there are only two case reports of dermatomyositis presenting with Degos-like lesions and two case reports of progressive systemic sclerosis in the English literature.Citation4–Citation7 Herein, we present two additional cases of Degos-like lesions associated with dermatomyositis and the first case of Degos-like lesions linked to systemic sclerosis overlapping SLE. We also performed the literature review on Degos-like lesions as a cutaneous manifestation of CTDs.
Case Reports
Case 1
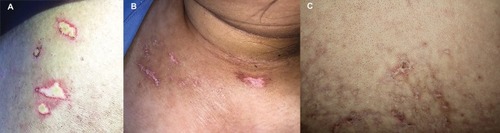
A 59-year-old woman diagnosed with cervical cancer 10 years ago and was successfully treated with brachytherapy. She presented with progressive proximal muscle weakness, dysphagia, and skin rash for 2 months. Dermatologic examination showed erythematous patches on the nose, scalp, and upper back, together with multiple small porcelain-white papules with peripheral telangiectasia on her back, measuring 2–4 mm in size ().
Figure 1 (A and B) Small porcelain-white papules and macules with peripheral telangiectasia. (C) Multiple atrophic porcelain-white macules with peripheral telangiectasia on reticulated erythematous to brownish background.
The laboratory data showed a serum creatinine kinase level of 2637 IU/L; anti-nuclear antibody (ANA) was positive (fine speckled 1:1280, anti-nRNP 1+, anti-Sm 1+). Myositis-specific autoantibodies, anticardiolipin, lupus anticoagulant, beta2-glycoprotein, and cryoglobulin were all negative. Complete blood count, coagulation profiles, electrolyte, renal, and thyroid functions were within the normal limits. Electromyography (EMG) showed irritative myopathic change in left quadriceps and myopathic change in deltoid and biceps, which were suggestive of inflammatory myopathy. Nerve conduction study (NCV) revealed no evidence of polyneuropathy. The quadricep muscle biopsy showed many vacuolated fibers with wiped out appearance, mild-to-moderate perifascicular atrophy, and the presence of perivascular cellular reaction consistent with dermatomyositis.
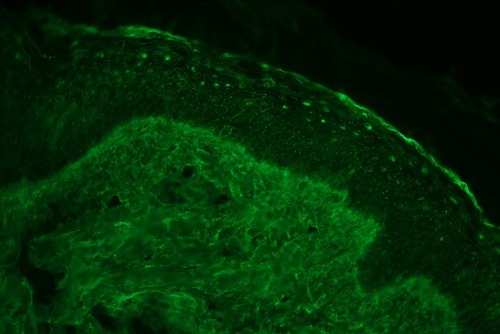
A skin biopsy on the atrophic white lesion revealed hyperkeratosis, epidermal atrophy, vacuolar alteration of basal keratinocytes. Marked papillary edema with extravasated red blood cells and telangiectasia (). Direct immunofluorescence showed epidermal nuclear staining ().
Figure 2 (A) Histopathology from case 1 showed hyperkeratotic epidermal atrophy, vacuolar alteration of basal keratinocytes, marked papillary edema with extravasated red blood cells, and telangiectasia (H&E, 100x). (B) Histopathology from case 3 showed wedge-shaped infarct in papillary dermis with sparse lymphoplasmacytic infiltrate and vacuolar alteration of basal cell layer. Homogenized eosinophilic altered collagen bundles within the entire dermis (H&E, 100x).
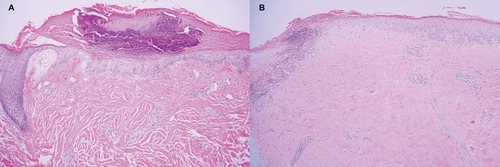
Figure 3 Direct immunofluorescence shows epidermal nuclear staining of immunoglobulin G (400x).
Intravenous immunoglobulin was given for 5 days, which resulted in remarkable improvement of the muscle strength as well as cutaneous lesions. The patient was later given methotrexate (7.5 mg/week) and high-dose systemic steroid (prednisolone 1 mg/kg/day), which was later maintained at 5–10 mg/day. Unfortunately, she was found to have recurrent cervical cancer stage IV with lung and liver metastasis and expired in 2 years after.
Case 2
A 19-year-old previously healthy woman had a rapidly progressive proximal muscle weakness for 1 week and developed itchy erythematous rashes on the chest, back, abdomen and lower legs. Cutaneous examination revealed multiple erythematous to violaceous reticulated patches with some hypopigmentation on chest and abdomen, several central porcelain-white macules with surrounding telangiectasia on V-shaped region of the neck and upper chest (), and erythematous patches with some excoriation on upper back and lateral aspect of both thighs resembling shawl sign and holster sign, respectively.
Laboratory evaluations revealed an elevated muscle enzyme creatine kinase of 34,950 IU/L, aspartate transaminase (AST) of 903 U/L, and alanine transaminase (ALT) of 274 U/L. Coagulation profiles, renal and thyroid function test results were unremarkable. Myositis-specific autoantibodies were all negative. Antinuclear antibody (ANA), anti-DNA, anticardiolipin, lupus anticoagulant, beta2-glycoprotein, and cryoglobulin were negative. EMG and NCV showed prominent active denervation in the distal and proximal muscles. The motor units showed myopathic features. Muscle biopsy demonstrated atrophic, vacuolated, degenerated, and necrotic fibers; presence of focal cellular reaction in endomysium and perivascular reaction in perimysium; together with widening of perimysial connective tissue with fragmentation and fibrosis. The clinical and laboratory findings were consistent with dermatomyositis.
Histological examination taken from porcelain-white macule showed epidermal hyperplasia, increased proliferation of blood vessels and fibroblast, no necrosis or fibrin deposit in vessel. Direct immunofluorescence was negative.
Pulse intravenous methylprednisolone was initiated for 3 days, followed by oral prednisolone 1 mg/kg/day and azathioprine 50 mg/day. The muscle strength and skin lesions gradually improved. Prednisolone was tapered and maintained at 15 mg/day.
Case 3
A 26-year-old male, presented with asymptomatic whitish and erythematous rashes on back for 3 months. He also complained of the hardening skin along the fingers and forearms and positive history of Raynaud’s phenomenon. He denied history of photosensitivity, arthralgia, weakness, weight loss, dyspnea, or dysphagia. Physical examination showed multiple atrophic white macules with peripheral telangiectasia on reticulated erythematous to brownish background located on the back (). He also had sclerodactyly and sclerotic skin from the fingers up to the elbows. Periungual erythema and ragged cuticles were presented. The abdomen was soft without hepatosplenomegaly. The heart and lungs were normal.
Laboratory data showed hemoglobin level of 13.8 g/dL, hematocrit of 40.7%, white blood cell count 3640/mm3 with 69% neutrophils, 26% lymphocytes, and 5% monocytes. Platelet count was 176,000/mm3. ANA test was positive >1:1280 in homogeneous and nucleolar pattern (anti-nRNP/Sm 3+, anti-Sm 2+, anti-Ro 3+, anti-Scl 70 3+). He had low complement levels, C3 level of 0.48 g/L (0.9–1.8 g/L), and C4 level of 0.07 g/L (0.1–0.4 g/L). Erythrocyte sedimentation rate (ESR) was 11 mm/hr. Antiphospholipid antibodies were negative. Serum creatinine was 0.6 mg/dL. His urinary analysis was normal.
Skin biopsy from atrophic white lesion on his back revealed wedge-shaped infarct in papillary dermis with sparse lymphoplasmacytic infiltrate and vacuolar alteration of basal cell layer. Homogenized eosinophilic altered collagen bundles were evident on the entire dermis ().
In view of the hematological and immunological findings, the patient fulfilled the Systemic Lupus Erythematosus International Collaborating Clinics (SLICC) criteria for SLE (absolute lymphocyte count < 1500/mm3, WBC < 4000/mm3, ANA positive, anti-Sm positive, low complement). The patient was diagnosed with diffuse systemic sclerosis based on cutaneous and serological findings. Further investigations on systemic sclerosis showed no abnormality of the chest films or high-resolution computerized tomography (HRCT) scan of the chest. Pulmonary function tests were normal in spirometry, lung volume, and diffusing capacity for carbon monoxide (DLCO). The echocardiogram showed normal ventricular function with left ventricular ejection fraction (LVEF) of 75% and normal mean pulmonary artery pressure.
The patient received cyclophosphamide 50 mg/day, prednisolone 30 mg/day, nifedipine 20 mg/day, hydroxychloroquine 200 mg/day, aspirin 81 mg/day, and colchicine 1.2 mg/day. After 4 weeks of treatment, the skin lesions gradually improved. Prednisolone was later tapered to 15 mg/day.
Discussion
Degos disease is a syndrome of vasculopathy characterized by pathognomonic cutaneous lesions. Three classic clinical stages of skin lesions have been described. First is the inflammatory phase demonstrating skin-colored to erythematous firm papules. In second phase, papules later turn into central necrotic lesions. For the resolution phase, typical lesions marked by porcelain-white scars with surrounding erythema. Lastly, only white scars with irregular border remain.Citation3,Citation8 The character of Degos-like lesions is indistinguishable from classic Degos disease.
There is a wide spectrum of histologic findings in Degos disease, depending on phase of the lesions. Early lesions may demonstrate lymphocytic perivascular infiltration, dermal mucin deposition, and vacuolar alteration with few necrotic keratinocytes along dermo-epidermal junction. Fully developed papules may show more prominent dermo-epidermal interface with melanin incontinent, epidermal atrophy, and dermal sclerosis. Other features include vasculitis and thrombotic vasculopathy. Late atrophic papules may reveal wedge-shaped necrosis of the papillary and upper reticular dermis, more prominent dermal sclerosis and sparse inflammation accompany thrombotic vasculopathy.Citation2,Citation4,Citation9
The etiology of Degos disease is still unclear. Suggested hypotheses are vasculitis, coagulopathy, and primary dysfunction of the endothelial cells. Recent report suggests that C5b-9 deposition in vessels and the presence of stromal cell-derived factor-1/CXCL12 in the inflammatory cells of Degos’ patients also play a major role in the pathogenesis.Citation10 These factors may contribute to microvascular injury of the cutaneous arterioles and venules.
Degos disease can be classified into two subtypes.Citation9 Firstly, classic Degos disease which is characterized by typical skin lesion and internal organ involvement, mainly gastrointestinal tract and central nervous system. Cardiovascular and genitourinary system involvement have been rarely reported. However, up to 15% of classic Degos disease has skin limited presentation.Citation2,11 Secondly, Degos-like lesions which are most frequently associated with CTDs. Despite the clinical differences between classic Degos disease and cutaneous manifestations of CTDs, underlying vasculopathy may be a shared pathogenesis among both conditions.
summarizes the clinical, laboratory, histopathology, direct immunofluorescent, treatment, and prognosis of Degos-like lesions in association with CTDs reported in the English literature. In view of Degos-like lesions in CTDs, the age of affecting patients range from 19 to 59 years old and the common age group were young adults with mean age of 30.9 years old (18–35 years old). There is a known female predilection in CTDs. Likewise, in Degos-like lesions, in this population were predominantly females with a male to female ratio of approximately 1:2. However, all reported cases of systemic sclerosis were male patients. The location of the Degos-like skin lesions secondary to CTDs did not differ from classic Degos disease. The trunk and extremities were the most commonly involved areas. Although there were some reports on the connection between antiphospholipid antibodies and Degos disease,Citation12 our literature review reveals that most patients with Degos-like lesions in CTDs lacked antiphospholipid antibodies. With regards to systemic involvement, 2 out of 11 SLE patients with Degos-like lesions had major internal lupus, comprising renal and central nervous system involvement.Citation13,Citation15 In dermatomyositis, 3 patients had severe muscle involvement.Citation5 According to previous reports on Degos-like lesions in association with progressive systemic sclerosis, all patients had pulmonary and cardiac involvement.Citation6,Citation11 One patient died of renal crisis in 3 months. In all CTDs with Degos-like lesions, there were no gastrointestinal symptoms as typically seen in classic Degos disease.
Table 1 Demographic Data, Clinical, Laboratory, Histopathologic, Immunopathologic Characteristics, Treatment, And Outcome Of Degos-Like Lesions In Patients With Connective Tissue Diseases
Our first two cases presented with severe proximal muscle weakness but without pathognomonic cutaneous signs of dermatomyositis. As there were typical findings on EMG consistent with inflammatory myopathy and muscle biopsy showed prominent inflammation in perimysium and perivascular area consistent with inflammatory myositis, the results were compatible with dermatomyositis. It is noteworthy to emphasize that in both our dermatomyositis cases associated with Degos-like lesions and two other reported cases, all had negative myositis-specific autoantibody profiles.Citation4,Citation5 Moreover, Degos-like lesions may be a marker of systemic changes. In a report by Tsao et al, the appearance of Degos-like lesions signaled worsening of the weakness in a patient with dermatomyositis.Citation5
In our final case of Degos-like lesions that occurred in a patient overlapping systemic sclerosis and SLE. Unlike previous reports, thus far, despite extensive investigations, this patient lacked major internal organ involvement. However, close and active follow-up on internal organ involvement is mandatory for this patient.
Histopathologic study of Degos-like lesion in our cases showed findings compatible with both CTDs and Degos disease. These features support the hypothesis that Degos-like lesions may be not a specific entity but can be considered as a unique clinical and histological pattern expressed in patients with CTDs.Citation4,Citation9
With respect to Degos-like lesion and active/relapse disease, the lesions appear as relapse of cancer in one of our patients. It was also a marker for severe myopathy in both cases of dermatomyositis. However, due to its rarity, there are limited data on whether or not Degos-like lesions secondary to CTDs hold a prognostic value for recurrence or active underlying disease. Future studies are warranted.
Conclusions
Degos-like lesions are mostly associated with connective tissue diseases. They are characterized by atrophic porcelain-white papules with peripheral telangiectasia, identical to classic Degos disease. We report two cases of Degos-like lesions in association with dermatomyositis with severe muscle involvement and negative myositis-specific autoantibody profiles. We also present the first case of Degos-like lesions in diffuse systemic sclerosis overlapping SLE with no major organ involvement. The skin lesions gradually improved after aggressive treatment of the underlying CTDs themselves.
Ethical Statement
Written informed consent was provided by the patients (and the next of kin in case 1) to have their case details and images published. Institutional approval was not required to publish the case details.
Disclosure
The authors report no conflicts of interest in this work.
References
- Pirolla E, Fregni F, Miura IK, Misiara AC, Almeida F, Zanoni E. Degos disease - malignant atrophic papulosis or cutaneointestinal lethal syndrome: rarity of the disease. Clin Exp Gastroenterol. 2015;8:141–147. doi:10.2147/CEG.S5979425926751
- Kim E, Motaparthi K. Benign atrophic papulosis (Degos Disease) with lymphocytic vasculitis and lichen sclerosus-like features. Am J Dermatopathol. 2018;40(4):272–274. doi:10.1097/DAD.000000000000084728244939
- Scheinfeld N. Malignant atrophic papulosis. Clin Exp Dermatol. 2007;32(5):483–487. doi:10.1111/j.1365-2230.2007.02497.x17692056
- High WA, Aranda J, Patel SB, Cockerell CJ, Costner MI. Is Degos’ disease a clinical and histological end point rather than a specific disease? J Am Acad Dermatol. 2004;50(6):895–899. doi:10.1016/j.jaad.2003.11.06315153891
- Tsao H, Busam K, Barnhill RL, Haynes HA. Lesions resembling malignant atrophic papulosis in a patient with dermatomyositis. J Am Acad Dermatol. 1997;36(2 Pt 2):317–319. doi:10.1016/S0190-9622(97)80407-09039209
- Durie BGM, Stroud JD, Kahn JA. Progressive systemic sclerosis with malignant atrophic papulosis. JAMA Dermatol. 1969;100(5):575–581. doi:10.1001/archderm.1969.01610290059012
- Liu CM, Harris RM, Hansen CD. Lesions resembling malignant atrophic papulosis in a patient with progressive systemic sclerosis. Cutis. 2005;75(2):101–104.15773530
- Jang MS, Park JB, Yang MH, et al. Degos-like lesions associated with systemic lupus erythematosus. Ann Dermatol. 2017;29(2):215–218. doi:10.5021/ad.2017.29.2.21528392651
- Ball E, Newburger A, Ackerman AB. Degos’ disease: a distinctive pattern of disease, chiefly of lupus erythematosus, and not a specific disease per se. Am J Dermatopathol. 2003;25(4):308–320. doi:10.1097/00000372-200308000-0000512876488
- Theodoridis A, Makrantonaki E, Zouboulis CC. Malignant atrophic papulosis (Kohlmeier-Degos disease) - a review. Orphanet J Rare Dis. 2013;8:10. doi:10.1186/1750-1172-8-1023316694
- Coskun B, Saral Y, Cicek D, Ozercan R . Benign cutaneous Degos’ disease: a case report and review of the literature. J Dermatol. 2004;31(8):666–670. doi:10.1111/j.1346-8138.2004.tb00574.x15492441
- Harvell JD, Williford PL, White WL. Benign cutaneous Degos’ disease: a case report with emphasis on histopathology as papules chronologically evolve. Am J Dermatopathol. 2001;23(2):116–123. doi:10.1097/00000372-200104000-0000611285406
- Dubin HV, Stawiski MA. Systemic lupus erythematosus resembling malignant atrophic papulosis. Arch Intern Med. 1974;134(2):321–323. doi:10.1001/archinte.1974.003202001310194843199
- Black MM, Hudson PM. Atrophie blanche lesions closely resembling malignant atrophic papulosis (Degos’ disease) in systemic lupus erythematosus. Br J Dermatol. 1976;95(6):649–652. doi:10.1111/j.1365-2133.1976.tb07040.x1009012
- Doutre MS, Beylot C, Bioulac P, Busquet M, Conte M. Skin lesion resembling malignant atrophic papulosis in lupus erythematosus. Dermatologica. 1987;175(1):45–46. doi:10.1159/000248781
- Torok L. Symptomatic atrophic papulosis in a patient with systemic lupus erythematosus. Clin Exp Dermatol. 1996;21(5):390–392. doi: 10.1111/j.1365-2230.1996.tb00135.x
- Chan N, CY Leung. Malignant atrophic papulosis (Degos’ disease). Hong Kong J Dermatol Venereol. 2008;16:27–33.
- Riyaz N, Saleem R, Shafeeq R. Degos disease-like presentation in systemic lupus erythematosus. Indian J Dermatol Venereol Leprol. 2011;77(2):219–221. doi:10.4103/0378-6323.7747721393965
- Stephansson EA, Niemi KM, Jouhikainen T, Vaarala O, Palosuo T. Lupus anticoagulant and the skin. A longterm follow-up study of SLE patients with special reference to histopathological findings. Acta Derm Venereol. 1991;71(5):416–422.1684471