
Abstract
Castleman disease (CD) is a rare heterogeneous lymphoproliferative disorder and presents as unicentric Castleman disease (UCD) or multicentric Castleman disease (MCD). Cutaneous manifestations of MCD are rare and variable. Here, we reported an unusual case of MCD that initially presented with localized plaques on the scalp.
Keywords:
Introduction
Castleman disease, first described in 1954 by Castleman,Citation1 is a rare heterogeneous lymphoproliferative disorder. CD includes unicentric CD (UCD) and multicentric CD (MCD). Cutaneous manifestations of MCD are rare and variable. Paraneoplastic pemphigus is the most common cutaneous manifestation, and papules, plaques, and nodules are the second most frequent.Citation2 Here, we reported a case of MCD with rare cutaneous manifestation as its initial presentation.
Case Report
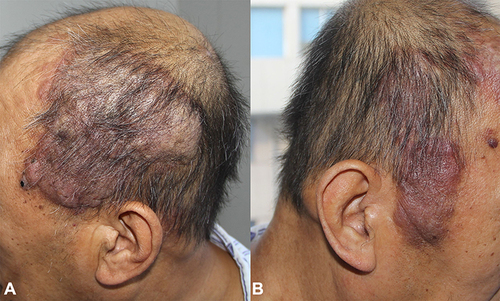
A 57-year-old man was referred to our hospital with complaints of multiple dark red nodules on the scalp for five years. Five years ago, a soy-sized nodule appeared on the scalp without any discomfort and gradually increased in number and size. The patient was sent to the local hospital seven months ago because of blood in the sputum. The biopsy of the lymph nodes behind the left ear showed that it was in line with Castleman disease, plasma cell type (IgG4>50/HPF, IgG4/IgG>40%). Computed tomography (CT) revealed multiple lymphadenopathies, including the right heart-phrenic angle, retroperitoneum, bilateral iliac area, and groin. Bone marrow biopsy was normal. He was diagnosed with Castleman disease (multicentric plasma cell type). Physical examination revealed multiple irregular-shaped, dark-red, soft to fluctuant, well-demarcated plaques on the scalp ranging from 1 to 8 cm in diameter ().
Figure 1 (A and B) Multiple irregular-shaped, dark-red, soft to fluctuant, well-demarcated, and infiltrated plaques ranging in size from 1 to 8 cm in diameter disseminating on the head.
Laboratory tests showed a decreased hemoglobin (Hb, 89 g/L; reference range [RR], 115–150 g/L), elevated erythrocyte sedimentation rate (ESR, 111 mm/h; RR, <20 mm/h), increased C-reactive protein (CRP, 70.4 mg/L; RR, 0–8 mg/L) and positive rheumatoid factor (RF, 21.2IU/mL; RR, 0–15 IU/mL). Serum IgG and IgA levels were high (respectively 62.6 g/L; RR, 7.2–16.8 g/L, 8.90 g/L; RR, 0.82–4.53 g/L), whereas IgM level was normal (1.68 g/L; RR, 0.46–3.04 g/L). Protein immunofixation of serum and urine were negative for M-protein. The serum total protein level was increased (99.9g/L; RR, 60–80 g/L), while the albumin decreased (24.2 g/L; RR, 34–48 g/L), and the albumin-globulin ratio was 0.82 (RR, 1–2). Serum levels of IgG4 (943.0mg/dl; RR, 3–201 mg/dl) were also increased. Serum levels of kappa (49.3g/L g/L; RR, 1.7–3.7 g/L) and lambda (23.4g/L; RR, 0.9–2.10g/L) light chain and urine levels of kappa (34.90mg/dL; RR, 1.7–3.7 g/L) and lambda (12.10mg/dL; RR, 0.9–2.10g/L) light chain was all significantly increased. Serum levels of VEGF (264.21pg/mL; RR, 0–142pg/mL) and interleukin-6 (IL-6, 50.23pg/mL; RR<5.9pg/mL) were all markedly increased. Serologic tests for hepatitis virus types B and C, HHV-8, CMV, EBV, ADV, tuberculosis, syphilis, and HIV antibody were all negative. The biopsy of the lymph nodes behind the left ear showed that small lymphocytes of the mantle zones are arranged in concentric rings around the atrophied germinal center, and amounts of plasma cells in the interfollicular region. The immunohistochemistry results are as followed: CD3 (partial +), CD20 (partial +), Ki67 (+), CD38 (+), CD138 (+), Kappa (+), Lambda (+), BCL-2 (perifollicular +), CD21 (dendritic cell+), CD34 (vascular+), IgG (+), IgG4 (+) (). The skin biopsy taken from the right ear revealed variable-sized lymphoid follicles, infiltrated neutrophils, and many plasma cells in the dermis. A broad immunohistochemical panel of CD3, CD20, CD38, CD138, IgG, IgG4, CD34, Kappa, Lambda, BCL-2, and Ki-67 was performed (). Based on the clinical, laboratory, and pathological findings, the patient was diagnosed with the plasmacytic variant of idiopathic multicentric CD (pMCD). The patient was treated with bortezomib, cyclophosphamide, and dexamethasone, and the cutaneous lesions disappeared after two months.
Figure 2 (A) Histopathology showed that small lymphocytes of the mantle zones are arranged in concentric rings around the atrophied germinal center, and amounts of plasma cells in the interfollicular region (HE, A×100). The immunohistochemistry results of CD3 (a, ×40), CD20 (b, ×40), Ki67 (c, ×40), CD38 (d, ×40), CD138 (e, ×40), Kappa (f, ×40), Lambda (g, ×40), BCL-2 (h, ×40), CD21 (i, ×40), CD34 (j, ×40), IgG (k, ×40), IgG4 (l, ×40).
Figure 3 (A and B), (A) showed reactive lymphoid follicular hyperplasia within the dermis. (B) revealed neutrophil and plenty of plasma cells infiltration. (HE, A×40, B×200). The immunohistochemistry results of CD3 (a, ×40), CD20 (b, ×40), CD38 (c, ×40), CD138 (d, ×40), IgG (e, ×40), IgG4 (f, ×40), CD34 (g, ×40), Kappa (h, ×40), Lambda (i, ×40), BCL-2 (j, ×40) and Ki-67 (k, ×40).
Discussion
CD is considered as an orphan disease with a prevalence of 21–25 cases per million person.Citation3 According to the Castleman Disease Collaborative Network (CDCN),Citation4 MCD includes idiopathic MCD (iMCD), human herpes virus-8 associated MCD (HHV8-MCD), and POEMS-MCD (polyneuropathy, organomegaly, endocrinopathy, monoclonal plasma cell disorder, skin changes). iMCD could be further subclassified into iMCD-NOS (iMCD–not otherwise specified) and iMCD-TAFRO (thrombocytopenia, ascites, reticulin fibrosis, renal dysfunction, organomegaly).
Skin abnormalities including paraneoplastic pemphigus, papules, plaques, nodules, xanthogranulomas, hyperpigmentation, cherry hemangiomatosis, and Kaposi sarcoma, though rare, have also been reported.Citation2,Citation5 However, MCD with localized dark-red papules as the initial presentation has rarely been reported. Chavez-Alvarez et analyzed 68 cases of CD and found that only 10 patients presented with localized plaques, papules, or nodules. The most frequent locations for the lesions are the trunk and extremities, while the scalp is rarely affected.Citation2
The pathogenesis of CD remained unclear. One probable theory was that HHV-8 might promote differentiation of naive B cells into plasmablasts without a germinal center reaction and produce a viral homolog of interleukin-6, which was responsible for symptoms.Citation6 Diagnosis usually imposes a challenge to CD due to the variable manifestations. A definite diagnosis is made by lymph node biopsy since skin biopsies are not diagnostic. CD usually presents with three histologic variants—hyaline vascular (or hypervascular), plasma cell (or plasmacytic), and mixed subtype.Citation6 The hyaline vascular subtype has small germinal centers and mantle lymphocytes surrounded by blood vessels and sclerotic collagen. The plasma cell subtype is multicentric and, the mixed subtype presents features of both.Citation7
The differential diagnosis of iMCD is broad. Multiple autoimmune diseases, such as rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), and Immunoglobulin-G4–related disease (IgG4-RD), as well as acute infections and malignancies, are all included. IgG4-RD is a fibroinflammatory condition that has distinctive clinical findings, involves multiple organs, and has unique histopathology-a lymphoplasmacytic infiltrate, obliterative phlebitis, fibrosis, and modest tissue eosinophilia,Citation8 but little was reported about its cutaneous manifestations. In our case, obliterative phlebitis, fibrosis, and modest tissue eosinophilia were absent, parotid gland and salivary gland were not involved, although serum levels of IgG4 were increased. A discussion about the overlap between IgG4-RD and iMCD led to the consensus that iMCD should supersede a diagnosis of IgG4-RD, even with very high IgG4 levels.Citation9,Citation10
Due to the rarity of the disease, there are no clinical studies available comparing the effect of different treatment modalities such as chemotherapy, rituximab, and anti-IL6 mAbs. Treatment for patients with MCD is based on the severity of the disease. There are consensus treatment recommendations for MCD.Citation11
Conclusion
Here we present an unusual case of localized plaques on the scalp in a MCD patient. Localized plaques of MCD, though rare, have also been reported. However, MCD with localized dark-red plaques as the initial presentation has rarely been reported. Clinicians should be aware that skin abnormalities of MCD may occur before the system involvement.
Ethics and Consent Statements
Written informed consent was provided by the parent of the patient to have the case details and any accompanying images published. Institutional approval was not required to publish the case details.
Disclosure
The authors report no conflicts of interest in this work.
Additional information
Funding
References
- Castleman B, Iverson L, Menendez V. Localized mediastinal lymphnode hyperplasia resembling thymoma. Cancer. 1956;9(4):822–830. doi:10.1002/1097-0142(195607/08)9:4<822::AID-CNCR2820090430>3.0.CO;2-4
- Chavez-Alvarez S, Villarreal-Martinez A, Ocampo-Candiani J, et al. Cutaneous manifestations of Castleman disease. Int J Dermatol. 2020;59(10):1226–1240. doi:10.1111/ijd.15043
- Wu D, Lim M, Jaffe E. Pathology of Castleman disease. Hematol Oncol Clin North Am. 2018;32(1):37–52. doi:10.1016/j.hoc.2017.09.004
- Fajgenbaum D, Uldrick T, Bagg A, et al. International, evidence-based consensus diagnostic criteria for HHV-8-negative/idiopathic multicentric Castleman disease. Blood. 2017;129(12):1646–1657. doi:10.1182/blood-2016-10-746933
- Kim HJ, Han JH, Bang CH, et al. Cutaneous disorders associated with Castleman’s disease. Acta Derm Venereol. 2019;99(11):984–989. doi:10.2340/00015555-3253
- Dispenzieri A, Fajgenbaum D. Overview of Castleman disease. Blood. 2020;135(16):1353–1364. doi:10.1182/blood.2019000931
- Okuyama R, Harigae H, Moriya T, et al. Indurated nodules and plaques showing a dense plasma cell infiltrate as a cutaneous manifestation of Castleman’s disease. Br J Dermatol. 2007;156(1):174–176. doi:10.1111/j.1365-2133.2006.07577.x
- Khosroshahi A, Stone J. A clinical overview of IgG4-related systemic disease. Curr Opin Rheumatol. 2011;23(1):57–66. doi:10.1097/BOR.0b013e3283418057
- Deshpande V, Zen Y, Chan J, et al. Consensus statement on the pathology of IgG4-related disease. Mod pathol. 2012;25(9):1181–1192. doi:10.1038/modpathol.2012.72
- Umehara H, Okazaki K, Masaki Y, et al. Comprehensive diagnostic criteria for IgG4-related disease (IgG4-RD), 2011. Mod Rheumatol. 2012;22(1):21–30. doi:10.3109/s10165-011-0571-z
- van Rhee F, Voorhees P, Dispenzieri A, et al. International, evidence-based consensus treatment guidelines for idiopathic multicentric Castleman disease. Blood. 2018;132(20):2115–2124. doi:10.1182/blood-2018-07-862334