Abstract
Given that biosimilars are agents that are similar but not identical to the reference biopharmaceutical, this study aims to introduce and describe specific issues related to the economic evaluation of biosimilars by focusing on the relative costs, relative effectiveness, and cost-effectiveness of biosimilars. Economic evaluation assesses the cost-effectiveness of a medicine by comparing the costs and outcomes of a medicine with those of a relevant comparator. The assessment of cost-effectiveness of a biosimilar is complicated by the fact that evidence needed to obtain marketing authorization from a registration authority does not always correspond to the data requirements of a reimbursement authority. In particular, this relates to the availability of adequately powered equivalence or noninferiority studies, the need for comparative data about the effectiveness in a real-world setting rather than the efficacy in a structured setting, and the use of health outcome measures instead of surrogate endpoints. As a biosimilar is likely to be less expensive than the comparator (eg, the reference biopharmaceutical), the assessment of the cost-effectiveness of a biosimilar depends on the relative effectiveness. If appropriately designed and powered clinical studies demonstrate equivalent effectiveness between a biosimilar and the comparator, then a cost-minimization analysis identifies the least expensive medicine. If there are differences in the effectiveness of a biosimilar and the comparator, other techniques of economic evaluation need to be employed, such as cost-effectiveness analysis or cost-utility analysis. Given that there may be uncertainty surrounding the long-term safety (ie, risk of immunogenicity and rare adverse events) and effectiveness of a biosimilar, the cost-effectiveness of a biosimilar needs to be calculated at multiple time points throughout the life cycle of the product.
Introduction
Biopharmaceutical medicines are reference or originator medicinal products made by or derived from living organisms using biotechnology ().Citation1 Biotechnology refers to the use of biological systems (eg, bacteria, yeast, and human cells) to identify, sequence, and manipulate DNA aimed at producing therapeutic and medical diagnostic products.Citation2 The class of biopharmaceuticals has been available for more than 20 years and includes blood coagulation modulators, enzymes, erythropoietins, gonadotrophins, granulocyte colony-stimulating factors (G-CSFs), human growth hormones, human insulins, interferons, interleukins, monoclonal antibodies, tissue plasminogen activators, and vaccines. Biopharmaceuticals tend to have a large size and a complex structure and are manufactured from a unique line of living cells, making it impossible to ensure an identical copy. This contrasts with chemical medicines, which tend to have a small size and simple structure and are manufactured using a predictable chemical process that generates identical copies.
Figure 1 Outline of biotechnology medicines.
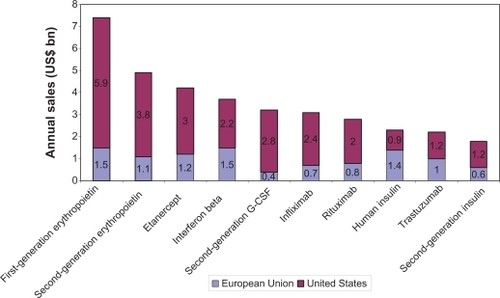
Biopharmaceuticals represent a fast-growing segment of the pharmaceutical market, constituting 32% of products in the development pipeline and 7.5% of marketed medicines and accounting for around 10% of pharmaceutical expenditure.Citation3 The annual sales of the top ten biopharmaceuticals in 2006 are illustrated in . The biopharmaceutical market is expected to grow exponentially at more than 20% per year, as a result of a burgeoning pipeline, approval for more common conditions, increased utilization, and expanding indications.Citation4 This growth can be exemplified by the market access of new biopharmaceuticals that target larger populations in the presence of competitor medicines (eg, insulins for diabetes mellitus affecting 194 million patients worldwide).Citation5
Figure 2 Top 10 biopharmaceuticals in sales in 2006.Citation41
In the European Union, the first patents on biopharmaceuticals expired in 2001, and the first biosimilar medicines or follow-on biologics were approved by European Medicines Agency in April 2006. To date, biosimilars of recombinant human erythropoietins (epoetin alfa and epoetin zeta), G-CSFs (filgrastim), and human growth hormones (somatropin) have entered the European market. In the coming years, patents will expire on some major biopharmaceuticals such as interferons and insulins. Probably, this will lead to the market entry of a number of biosimilars in the not too distant future.
Biosimilars are agents that are similar but not identical to the reference biopharmaceutical (). Therefore, a regulatory framework is in place in, for example, the European Union to assess the application for marketing authorization of biosimilars.Citation6 In addition, a regulatory framework has been introduced in the United States in 2010, although it is not clear how these rules will be implemented and how they will play out in practice.Citation7 In general, a biosimilar is registered if it is similar to the reference biopharmaceutical in terms of safety, quality, and efficacy. Dossiers of biosimilars tend to include data from clinical trials with a view to demonstrating similar safety and efficacy with the reference biopharmaceutical. In light of the variation between biotechnology medicines, the marketing authorization process is specific to each product. For instance, the European Medicines Agency has published additional guidelines that relate specifically to biosimilars containing monoclonal antibodiesCitation8 and biosimilars containing recombinant interferon β.Citation9
In addition to the factors mentioned above – the rapid growth of the biotechnology market, the imminent patent expiry on several major biopharmaceuticals, and the establishment of regulatory frameworks – the key driver for the biosimilar market is likely to be cost containment pressures in health care systems in the context of aging populations and of the current financial and economic crisis. For instance, the European Generic Medicines Agency has estimated that biosimilars generated annual savings of ∼€ 1.4 billion in the European Union in 2009.Citation10
Economic evaluation serves to guide the implementation of safe and cost-effective medicines that support further health improvements, while containing health expenditure. Economic evaluation assesses the cost-effectiveness of a medicine by comparing the costs and outcomes of a medicine with those of a relevant comparator.Citation11 Evidence derived from economic evaluations is used to inform pharmaceutical reimbursement decisions in many countries. The requirement for economic evaluation fits within an overall trend toward evidence-based decision-making in health care.Citation12
The results of an economic evaluation can be expressed in the form of an incremental cost-effectiveness ratio. This ratio relates the difference in costs between a medicine and the comparator to the difference in outcomes. The incremental cost-effectiveness ratio can be represented as a point on the cost-effectiveness plane ().Citation11 On the horizontal axis, the difference in effectiveness (eg, life years) between a medicine and the comparator is portrayed. The vertical axis represents the cost difference between a medicine and the comparator. A medicine may have higher or lower costs and higher or lower effectiveness than the comparator, so that its point may fall into one of the four quadrants.
Figure 3 The cost-effectiveness plane.
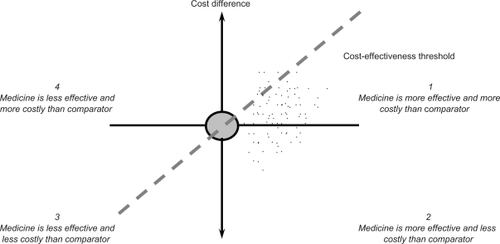
The incremental cost-effectiveness ratio can then be compared with a threshold incremental cost-effectiveness ratio, which reflects the maximum cost per unit of outcome that a health care payer is willing to pay for a medicine. This means that a medicine with an incremental cost-effectiveness ratio below the threshold value is likely to be accepted by a health care payer and a medicine with an incremental cost-effectiveness ratio exceeding the threshold value is likely to be refused. provides an overview of threshold incremental cost-effectiveness ratios in selected countries. Such threshold ratios are usually applied to medicines, but are relevant to any health technology. The gradient of the dashed line in represents a specific threshold incremental cost-effectiveness ratio. A medicine is cost-effective if its point estimate falls to the southeast of this dashed line.
Table 1 Threshold incremental cost-effectiveness ratios in selected countries
The aim of this study is to introduce and describe specific issues related to the economic evaluation of biosimilars. This study provides insight into the cost-effectiveness of biosimilars to academic researchers, pharmaceutical companies that set up biosimilar research and development programs, and policy makers who make decisions about reimbursement of biosimilars.
Methods
This study was based on a review of the international literature focusing on the relative costs, relative effectiveness, and cost-effectiveness of biosimilars. The literature review did not wish to identify and discuss all economic evaluations of biosimilars, but rather drew on published economic evaluations with a view to identify and illustrate the factors affecting the cost-effectiveness of biosimilars. As such, the literature review of economic evaluations was not systematic.
Relevant studies were identified by searching PubMed, Embase, Centre for Reviews and Dissemination databases (Database of Abstracts of Reviews of Effects, National Health Service Economic Evaluation Database, and Health Technology Assessments Database), Cochrane Database of Systematic Reviews, and EconLit up to November 2010. Additionally, the bibliography of included studies was checked for other relevant studies. Search terms included ‘biotechnology’, ‘biopharmaceutical’, ‘biosimilar’, ‘follow-on biologic’, ‘market access’, ‘research and development’, ‘registration’, ‘pricing’, ‘reimbursement’, ‘health economics’, ‘pharmaco-economics’, ‘economic evaluation’, and ‘cost-effectiveness’ alone and in combination with each other.
The literature search included articles published in peer-reviewed journals. Additionally, the relevant congress abstracts were identified by searching Outcomes Research Digest, an electronic database of abstracts presented at the conferences of the International Society of Pharmacoeconomics and Outcomes Research.
Results
Relative effectiveness
Relative effectiveness, in the context of an economic evaluation, refers to the differences in effectiveness between a biosimilar and the comparator. Registration authorities, such as the European Medicines Agency or the US Food and Drug Administration, approve the marketing authorization of a biosimilar based on the assumption that the biosimilar generates equivalent outcomes as the reference biopharmaceutical. What does equivalence mean for assessing relative effectiveness?
Biopharmaceuticals require multifaceted manufacturing and purification processes, and changes to the manufacturing process can result in differences in quality, safety, and efficacy (ie, ‘the process is the product’).Citation13 When the manufacturer of a biosimilar establishes its own manufacturing process, this process is unlikely to be 100% the same as the process of the reference biopharmaceutical.Citation6 Subtle differences arise because biotechnology medicines are derived from living organisms and some process features of the reference biopharmaceutical remain confidential even after patent expiry.Citation14 Current analytical techniques and clinical studies are not able to detect all potential differences in clinical outcomes between a biosimilar and the reference biopharmaceutical.Citation15 Although the risk of immunogenicity and rare adverse events in the long term is particularly relevant to biotechnology medicines, the time horizon of biosimilar studies submitted to registration authorities is usually not long enough to consider these potential effects. For instance, the European Medicines Agency guideline relating to somatropin biosimilars states that one-randomized controlled trial comparing the biosimilar and the reference biopharmaceutical for, at least, 6 months is required for marketing authorization.Citation16
This implies that, in practice, a biosimilar may have lower or equal effectiveness than the reference biopharmaceutical. The case can also arise where a biosimilar is more effective than the reference biopharmaceutical (a so-called ‘bio-better’ medicine).Citation17 This may result from the fact that the biopharmaceutical is developed using a 15- to 20-year-old manufacturing process, whereas the biosimilar manufacturer makes use of the most recent manufacturing processes.
Evidence needed to obtain marketing authorization from a registration authority does not always correspond to the data requirements of a reimbursement authority for a number of reasons. First, to substantiate the claim of equivalence between a biosimilar and the reference biopharmaceutical, there is a need for adequately powered equivalence or non-inferiority studies. Such studies are available for some, but not for all biosimilars. For instance, the European Medicines Agency has accepted evidence from pharmacokinetic and pharmacodynamic studies only (eg, for filgrastim and recombinant human insulins) in the absence of noninferiority clinical studies. In addition, the European Medicines Agency may allow the extrapolation of data to another indication of the reference biopharmaceutical without an evaluation of the biosimilar in this new patient population.Citation18
Second, as the cost-effectiveness of a biosimilar is calculated relative to a comparator, there is a need for comparative data. However, clinical trials used for registration purposes usually employ placebo as a comparator. In contrast, reimbursement authorities require that the biosimilar be compared to the current standard treatment (eg, the reference biopharmaceutical). An indirect comparison can be set up using the evidence from placebo-controlled trials of the biosimilar and placebo-controlled trials of the reference biopharmaceutical, but such comparisons have a lower methodological quality as a direct head-to-head clinical trial of the biosimilar and the reference biopharmaceutical.
Third, registration authorities demand clinical trials that demonstrate efficacy in a structured setting. However, reimbursement authorities require data on the effectiveness of a biosimilar in a real-world setting.Citation19 For instance, the imposition of strict patient inclusion/exclusion criteria in clinical trials of biosimilars or the enrollment of healthy volunteers in clinical trials of recombinant G-CSF biosimilars restricts the generalizability of health outcome results and limits the use of such data for reimbursement purposes.Citation18 In addition, differences in treatment regimens between those studied in clinical trials and those applied in daily clinical practice may have a clinically relevant impact on health outcomes.
Finally, it should be noted that clinical trials used for registration purposes may employ surrogate outcome measures. For instance, clinical trials have measured the impact of an epoetin alfa biosimilar on patient hemoglobin levels.Citation20 In contrast, reimbursement authorities wish to have data about primary health outcomes, such as mortality or quality of life. To address this issue, health-economic modeling approaches can be employed if there is evidence of the relationship between the surrogate endpoint and the health outcome.
Relative costs
The relative costs or the cost difference between a biosimilar and the comparator in an economic evaluation depends on the cost of the medicines and other costs associated with biotechnology therapy. From a theoretical perspective, the relative costs should reflect the difference in opportunity costs (ie, the cost related to the next-best choice available with limited resources) between a biosimilar and the comparator. However, in practice, relative costs refer to the difference in medicine acquisition prices. On the one hand, comparisons based on acquisition prices rather than costs could be misleading because, for example, a manufacturer who is currently charging a high price might be willing to reduce it substantially in the face of competition. On the other hand, differences in acquisition prices between a biosimilar and the comparator are relevant to many reimbursement authorities.
With respect to chemical medicines, differences in acquisition prices between originator and generic medicines of up to 80% have been observed in countries such as Germany, the United Kingdom, and the United States.Citation21,Citation22 However, the price differential between biosimilars and biopharmaceuticals is likely to be smaller than that observed between originator and generic chemical medicines, given that biosimilars incur higher research and development costs. The developmental time for a generic medicine is around 3 years, whereas this period increases from 6 to 9 years for a biosimilar.Citation23 Generic medicines need to demonstrate bioequivalence only, whereas biosimilars need to conduct phase I and III clinical trials. Although there is no need to repeat all trials of the reference biopharmaceutical, the need to conduct some biosimilar trials enrolling several hundreds of patients involve considerable expense and time: a US study has estimated that the costs of biosimilar trials would range from US$10 to $40 million.Citation24 This study also reported that the required investment in biosimilar manufacturing processes amounts to US$250–$450 million. Furthermore, pharma-covigilance programs are usually instituted to follow up safety and efficacy of a biosimilar once the product has entered the market, thereby increasing the prices further. Differences in the acquisition price between a biosimilar and the reference biopharmaceutical in the region of 15%–30% have been suggested in the literature.Citation3,Citation23,Citation25 This price differential can be substantial when applied to expensive biopharmaceuticals, and it can be expected to increase as the acquisition price of biosimilars falls as they gain market share.Citation26 For instance, it has been estimated that a 20% price reduction of five off-patent biopharmaceuticals would save the European Union more than €1.6 billion per year.Citation27
Hospitals, the setting in which biosimilars tend to be prescribed, are likely to negotiate discounts on official medicine prices. In other words, competition between manufacturers takes the form of discounting to the distribution chain rather than price competition. No data on discounts for biosimilars are publicly available, but some studies have investigated discounting in the sector of generic chemical medicines. This research indicated that generic medicine discounts ranged from 20% to 70% off the wholesaler selling price in France, and maximum discounts exceeded 50% of the drug tariff price in the United Kingdom.Citation28,Citation29 Competition by discount may financially benefit hospitals, but is not sustainable in the long run, as health care payers and patients are likely to capture only some of the potential savings from a biotechnology medicines market where companies compete on price. In addition, as economic evaluations draw on official prices of the biosimilar and the biopharmaceutical, the calculated relative costs do not correspond with actual differences in the acquisition costs, and the cost-effectiveness of biosimilars is not calculated correctly.
Any potential differences in the (long-term) safety and effectiveness of a biosimilar and the reference biopharmaceutical may impose the need for additional health care, and generate health care costs and costs of productivity loss. This, in turn, is likely to influence the cost-effectiveness of a biosimilar.
Cost-effectiveness
An economic evaluation relates the relative costs of a medicine and the current standard treatment to their relative effectiveness.Citation11 In some cases, this means that the cost-effectiveness of a biosimilar needs to be established vis-à-vis the reference biopharmaceutical. In other cases, biosimilars have been developed for older biopharmaceuticals, for which second-generation biopharmaceuticals are now marketed and have become the standard treatment (eg, second-generation erythropoietins and second-generation G-CSFs).Citation3 This implies that the cost-effectiveness of the first-generation biosimilar needs to be determined relative to the second-generation biopharmaceutical.
This second case can be illustrated with the example of filgrastim for preventing febrile neutropenia. Filgrastim, the reference biopharmaceutical, has been marketed in the European Union since 1991, and five filgrastim biosimilars have entered the market since 2008 for the same indication. A long-acting pegylated form of filgrastim, pegfilgrastim, was registered by the European Medicines Agency in 2002. As pegfilgrastim has become the standard treatment to prevent febrile neutropenia, any economic evaluation of a filgrastim biosimilar should calculate its cost-effectiveness relative to pegfilgrastim. Assuming that the filgrastim biosimilar and biopharmaceutical have equal effectiveness, the requirement to use pegfilgrastim as comparator may negatively influence the cost-effectiveness of a filgrastim biosimilar, given that pegfilgrastim has been shown to be at least as effective as the filgrastim biopharmaceutical.Citation30
If clinical studies demonstrate an equal effectiveness profile of a biosimilar and the comparator, then a cost-minimization analysis needs to be carried out and the least expensive medicine is chosen. In , this means that the point estimate for cost-effectiveness is situated on the vertical axis where there is no difference in the effectiveness between the biosimilar and the comparator.
Cost-minimization analyses have been submitted to reimbursement authorities for biosimilars of epoetin alfa, filgrastim, and somatropin in the European Union. For instance, the Scottish Medicines Consortium approved the use of epoetin zeta (Retacrit®; Hospira UK Limited, Royal Leamington Spa, UK), a biosimilar to epoetin alfa, for the treatment of anemia associated with chronic renal failure in 2008.Citation20 As two phase III trials showed clinical equivalence for epoetin zeta when compared with epoetin alfa for the surrogate endpoints of correction and maintenance of hemoglobin concentration, the economic evaluation took the form of a cost-minimization analysis. The evaluation compared epoetin zeta with three other erythropoiesis-stimulating agents and concluded that epoetin zeta would yield equivalent efficacy at similar or lower costs.
The Scottish Medicines Consortium also accepted a filgrastim biosimilar (Ratiograstim®; Ratiopharm GmbH, UIm, Germany) for use within the National Health Service Scotland for the prevention of febrile neutropenia.Citation31 Equivalent efficacy of the biosimilar and the reference bio-pharmaceutical in terms of duration of severe neutropenia was supported by one randomized controlled trial in breast cancer patients. The manufacturer argued that the conclusions from the breast cancer trial were relevant to a range of different types of underlying diseases and to the performance of the biosimilar in general. The reimbursement application reported a cost-minimization analysis focusing on the costs of medicines and treatment of febrile neutropenia associated with the biosimilar and the reference biopharmaceutical. This analysis predicted savings with the biosimilar of £322 per patient over an 84-day period. Two analyses were conducted: the first used list prices of medicines and the second applied discounted prices. Finally, the Scottish Medicines Consortium remarked that no data had been submitted comparing the biosimilar with the second-generation biopharmaceutical, pegfilgrastim.
The National Institute for Health and Clinical Excellence recommended the use of a somatropin biosimilar to treat child growth deficiencies in England and Wales in May 2010.Citation32 Head-to-head trials demonstrated comparable efficacy of the biosimilar and the reference biopharmaceutical. In this case, where the two products are suitable, the Institute argued that the less expensive biosimilar should be chosen.
If there are differences in the effectiveness of a biosimilar and the comparator, other techniques of economic evaluation need to be employed, such as a cost-effectiveness analysis (eg, using life years as outcome measure) or a cost-utility analysis (eg, using quality-adjusted life years as outcome measure). Assuming that total costs of a biosimilar are lower than total costs of the comparator means that the point estimate for cost-effectiveness lies south of the horizontal axis in . The point estimate can fall in quadrant 2, where the biosimilar is more effective and less expensive than the comparator and, thus, is cost-effective. If the point estimate falls in quadrant 3, the biosimilar is less effective and less expensive than the comparator, and the incremental cost-effectiveness ratio needs to be calculated. In this case, the biosimilar is cost-effective when its point estimate falls to the southeast of the dashed line representing a specific threshold incremental cost-effectiveness ratio.
Given that no reimbursement authority has issued guidelines about which technique of economic evaluation is appropriate to calculate the cost-effectiveness of a biosimilar and given that there may be uncertainty surrounding relative effectiveness (cfr. supra), submissions to reimbursement authorities may include a cost-minimization analysis as well as a cost-effectiveness or cost-utility analysis.Citation15 Additionally, such exercises may carry out sensitivity analyses, exploring the impact of changes in relative effectiveness on the cost-effectiveness of a biosimilar.
Recently, innovative reimbursement mechanisms have been introduced by health care payers such as risk-sharing arrangements. A risk-sharing arrangement is a scheme in which the manufacturer shares the risk with the health care payer that the product may not be effective for a particular patient. If the product does not have the expected effect, the company may lose some or all product revenue or needs to provide a replacement product.Citation33 Such arrangements are instituted at the level of a defined patient population rather than a group of patients cared for by an individual institution or health care provider, may require physicians to be trained in the appropriate use of the product, and necessitate the implementation of a tracking system to follow up its use. As risk-sharing arrangements are in place for selected biop-harmaceuticals in some European countries, they are likely to be rolled out to apply to biosimilars as well and, thereby, influence their cost-effectiveness.Citation34
Due to potential concerns surrounding the long-term safety (ie, risk of immunogenicity and rare adverse events) and effectiveness of a biosimilar, there is a need to consider the cost-effectiveness of a biosimilar after a number of years following the admission to the reimbursement system in addition to the assessment of its cost-effectiveness at the time of the reimbursement application. Therefore, manufacturers need to explore setting up databases or observational studies to demonstrate the postlaunch cost-effectiveness of a biosimilar based on phase IV trials.
Conclusions
As a biosimilar is likely to be less expensive than the comparator (eg, the reference biopharmaceutical), the assessment of the cost-effectiveness of a biosimilar depends on the relative effectiveness. If appropriately designed and powered clinical studies demonstrate equivalent effectiveness between a biosimilar and the comparator, then a cost-minimization analysis needs to be carried out and the least expensive medicine is chosen. If there are differences in the effectiveness of a biosimilar and the comparator, other techniques of economic evaluation need to be employed, such as cost-effectiveness analysis or cost-utility analysis. Given that there may be uncertainty surrounding the long-term safety and effectiveness of a biosimilar, the cost-effectiveness of a biosimilar needs to be calculated at multiple time points throughout the life cycle of the product.
Disclosure
The author reports no conflicts of interest directly relevant to the content of this manuscript.
References
- NaglePCLugoTFNicitaCADefining and characterizing the late-stage biopharmaceutical pipelineAm J Manag Care20039Suppl 6S124S13514577717
- YoungFEBiotechnology: the view from the FDAHealth Matrix198643101510279339
- ZunigaLCalvoBBiosimilars–the way forwardHosp Pharm Europe2010503334
- CohenMMorrowTPennaPManaging the expanded use of biologics across therapeutic areas: an example from b-cell targeted therapiesAm J Manag Care200612Suppl 2S24S3716551202
- SimonFMarket access for biopharmaceuticals: new challengesHealth Aff (Millwood)20062551363137016966734
- European Medicines AgencyGuideline on Similar Biological Medicinal Products2006 Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003517.pdf. Accessed December 6, 2010
- United States CongressPatient Protection and Affordable Care Act2010 Available from: http://www.opencongress.org. Accessed December 6, 2010
- European Medicines AgencyGuideline on Similar Biological Medicinal Products Containing Monoclonal Antibodies2010 Available from http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/11/WC500099361.pdf. Accessed December 6, 2010
- European Medicines AgencyConcept Paper on Similar Biological Product Containing Recombinant Interferon BetaEuropean Medicines Agency2010 Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/04/WC500089210.pdf. Accessed December 6, 2010
- European Generic Medicines AssociationVision 2015. The EGA’s Thoughts on How to Improve the Legal and Regulatory Framework for Generic and Biosimilar Medicines2010 Available from: http://www.egagenerics.com/doc/EGA_Vision_2015.pdf. Accessed December 6, 2010
- DrummondMSculpherMJTorranceGWO’BrienBJStoddartGLMethods for the Economic Evaluation of Health Care Programmes3rd edOxford, UKOxford University Press2005
- PerlethMJakubowskiEBusseRWhat is ‘best practice’ in health care? State of the art and perspectives in improving the effectiveness and efficiency of the European health care systemsHealth Policy200156323525011399348
- MellstedtHNiederwieserDLudwigHThe challenge of biosimilarsAnn Oncol200819341141917872902
- RogerSDGoldsmithDBiosimilars: it’s not as simple as cost aloneJ Clin Pharm Ther200833545946418834359
- StewartAAubreyPBelseyJAddressing the health technology assessment of biosimilar pharmaceuticalsCurr Med Res Opin20102692119212620649394
- European Medicines AgencyAnnex Guideline on Similar Biological Medicinal Products Containing Biotechnology-Derived Proteins as Active Substance: Non-Clinical and Clinical Issues. Guidance on Similar Medicinal Products Containing Somatropin2006 Available from: http://www.ema.europa.eu/pdfs/human/biosimilar/9452805en.pdf. Accessed December 7, 2010
- De MoraFTorresRBiotechnology-derived medicines: what are they? A pharmacological and a historical perspectiveJ Generic Med201072145157
- HughesDABiosimilars: evidential standards for health technology assessmentClin Pharmacol Ther201087325726120160743
- HjelmgrenJBerggrenFAnderssonFHealth economic guidelines–similarities, differences and some implicationsValue Health20014322525011705185
- Scottish Medicines ConsortiumEpoetin Zeta Scottish Medicines Consortium; 2010. Available from: http://www.scottishmedicines.org.uk/smc/6094.html. Accessed December 7, 2010
- de JoncheereKRietveldAHHuttinCExperiences with genericsInt J Risk Safety Med200215101119
- KingDRKanavosPEncouraging the use of generic medicines: implications for transition economiesCroat Med J200243446246912187525
- MellstedtHThe future of biosimilarsHosp Pharm Europe2010493334
- GrabowskiHCockburnILongGThe market for follow-on biologics: how will it evolve?Health Aff (Millwood)20062551291130116966725
- LongMTroutJAkpinarPBiosimilars: HGH to TNFS, how will payers respondPriceSpective20091026
- European Generic Medicines AssociationEGA Handbook on Biosimilar Medicines2010 Available from: http://www.egagenerics.com/bio-handbook.htm. Accessed December 7, 2010
- OldhamTStrategies for entering the biosimilar marketOldhamTBiosimilars–Evolution or Revolution?London, UKBiopharm Knowledge Publishing2006
- KanavosPDo generics offer significant savings to the UK National Health Service?Curr Med Res Opin200723110511617257472
- KanavosPTaylorDPharmacy discounts on generic medicines in France: is there room for further efficiency savings?Curr Med Res Opin200723102467247617764613
- SienaSPiccartMJHolmesFAGlaspyJHackettJRenwickJJA combined analysis of two pivotal randomized trials of a single dose of pegfilgrastim per chemotherapy cycle and daily Filgrastim in patients with stage II–IV breast cancerOncol Rep200310371572412684649
- Scottish Medicines ConsortiumRatiograstim2009 Available from: http://www.scottishmedicines.org.uk/files/filgrastim_Ratiograstim_FINAL_Oct_2009_Revised_031109.doc_for_website.pdf. Accessed December 8, 2010
- National Institute for Health and Clinical ExcellenceHuman Growth Hormone (Somatropin) for the Treatment of Growth Failure in Children (Review of NICE Technology Appraisal Guidance 42)2010 Available from: http://guidance.nice.org.uk/TA188. Accessed December 8, 2010
- CookJPVernonJAManningRPharmaceutical risk-sharing agreementsPharmacoeconomics200826755155618563946
- ZaricGSXieBThe impact of two pharmaceutical risk-sharing agreements on pricing, promotion, and net health benefitsValue Health200912583884519490563
- GeorgeBHarrisAMitchellACost-effectiveness analysis and the consistency of decision making: evidence from pharmaceutical reimbursement in Australia (1991 to 1996)Pharmacoeconomics200119111103110911735677
- LaupacisAFeenyDDetskyASTugwellPXHow attractive does a new technology have to be to warrant adoption and utilization? Tentative guidelines for using clinical and economic evaluationsCMAJ199214644734811306034
- RafteryJReview of NICE’s recommendations, 1999–2005BMJ200633275521266126816735341
- Health Care Insurance BoardRichtlijnen voor farmaco-economisch onderzoek (Guidelines for Pharmaco-Economic Research)Amstelveen, The NetherlandsHealth Care Insurance Board1999
- PHARMACPrescription for Pharmacoeconomic Analysis: Methods for Cost-Utility AnalysisAuckland, NZPHARMAC2007
- GrosseSDAssessing cost-effectiveness in healthcare: history of the $50,000 per QALY thresholdExpert Rev Pharmacoecon Outcomes Res20088216517820528406
- IMS HealthMidas DatabaseLondon, UKIMS Health2008