Abstract
Alzheimer’s disease (AD) is the most common neurodegenerative disorder, originating sporadically in the population aged over 65 years, and advanced age is the principal risk factor leading to AD development. In spite of the large amount of research going on around the globe and all the information now available about AD, there is still no origin or triggering process known so far. Drugs approved for the treatment of AD include tacrine, donepezil, rivastigmine, galantamine, and memantine. These may delay or slow down the degenerative process for a while, but they can neither stop nor reverse its progression. Because that this might be due to a lack of effect of these drugs on degenerating neurons, even when they are able to potentiate the brain in nondegenerative conditions, we propose here an alternative therapy consisting of initial repair of neuronal membranes followed by conventional drug therapies. The rehabilitation of neurons in a degeneration process would enable the drugs to act more effectively on them and improve the effects of treatment in AD patients.
Keywords:
Introduction
Alzheimer’s disease (AD) is a neurodegenerative disorder with loss of memory and other cognitive functions which progresses slowly, extending the disease for several years. Survival is very variable in these patients, although death often occurs within 10 years of onset, often as a result of infections.
In more than 90% of cases, AD develops after the age of 65 years, and doubles its prevalence with every successive decade of life, from 10% at 60–70 years to 40% at 80 years of age. Several chromosomes have been shown to be implicated in the pathology of AD, including chromosomes 1, 14, and 21, associated with the familial early-onset forms of the disease, and chromosomes 12 and 19, linked to late-onset forms.Citation1 However, most cases cannot be explained genetically, and thus several hypotheses have been raised over the years in an attempt to explain this complex disease, some of them suggesting the presence of unidentified infectious or toxic agentsCitation2 which modify the risk of developing AD.
One of the main processes leading to the symptomatology of AD is the abnormal phosphorylation of tau, a microtubule-associated protein. When tau gets hyperphosphorylated, it dissociates from microtubules, and structures called paired helical filaments begin to form within the cell bodies of neurons, generating neurofibrillary tangles, with hyperphosphorylated tau as their main component and leaving a loose cytoskeleton, which leads the neuronal membrane to lose its shape.
According to the amyloid cascade hypothesis,Citation3 β-amyloid peptide, the principal constituent of neuritic plaques, may have a major role in the neuropathology of AD. β-amyloid peptide generally exists in two forms, one is 40 amino acids long and the other is 42 amino acids long (Aβ40 and Aβ42 respectively), which differ in their terminal carbon structure. Of these two forms, Aβ42 is the most prone to aggregation.Citation4 Amyloid peptides are generated by the sequential actions of two enzymes, ie, β-secretase (BACE1) and γ-secretase.
BACE1 is widely expressed in the brain (mainly in the hypothalamus), and its involvement in the myelination process of the peripheral nervous system has been established.Citation5 Even though the function of BACE1 is not fully understood as yet, recent studies demonstrate that it activates neuroregulin-1, thus having direct involvement in the myelination process.Citation6 γ-secretase is a proteic presenilin complex, and one of its largest components is a protein known as nicastrin, the function of which has been widely associated with embryonic growth retardation and mortality.Citation7
There is no cure for AD as yet, and drug therapies are not effective enough to avoid symptoms. For these reasons, we felt the need to come up with an alternative, accessible, and safe therapy able to augment conventional drug treatments and have increased effectiveness in AD patients at any stage of the disease, but mainly in the first stages, where the rehabilitation of degenerating neurons could make a great difference to progression of the disease.
Drugs used for AD treatment
Five drugs have been approved for AD treatment in different stages of the disease, although they all show limited efficacy. These are tacrine, donepezil, rivastigmine, galantamine, and memantine.Citation8 Tacrine was one of the first drugs to be used for memory loss and cognitive decline, often accompanied by abnormal behavior and physical debilitation in AD patients. The alleged success of tacrine in treating these symptoms was heralded as confirmation of the cholinergic theory of AD. Nevertheless, its efficacy remains controversial.Citation9 A acetylcholinesterase inhibitor, tacrine has been associated with increased levels of transaminases in approximately 50% of the patients treated, but the mechanism by which it causes damage is not completely understood, and there could be genetic factors involved.Citation10
Donepezil is another acetylcholinesterase inhibitor its selectiveness as an inhibitor is fairly specific. Donepezil is used for the treatment of moderate to severe AD,Citation11 and is apparently well tolerated by patients and has few side effects.Citation12 On the other hand, rivastigmine has shown fewer side effects and better acceptance by patientsCitation13 than donepezil. However, the effectiveness of donepezil has been limited in patients with AD.Citation14
Galantamine, another member of the acetylcholinesterase inhibitor group, has shown a protective role for cortical neurons, preventing them from the cytotoxicity generated by aggregation of amyloid peptides.Citation15,Citation16
Unlike most drugs, memantine is not an acetylcholinesterase inhibitor, but an N-methyl-D-aspartate receptor antagonist. It reduces clinical deterioration in moderate to severe AD, for which other treatments are not available. Recent studies carried out in transgenic mice show that memantine decreases the levels of amyloid peptide and prevents synaptic dysfunction.Citation17 The activity of protein phosphatase (PP)-2A is compromised in the AD brain and is believed to be a cause of the abnormal hyperphosphorylation of tau and consequent neurofibrillary degeneration. In those studies, memantine inhibits and reverses the PP-2A inhibition-induced abnormal hyperphosphorylation and accumulation of tau in organotypic cultures of rat hippocampal slices. Such restorative effects of memantine were not detected either with 5,7-dichlorokynurenic acid or with D(-)-2-amino-5-phosphopentanoic acid, N-methyl-D-aspartate receptor antagonists, which are active at glycine and glutamate binding sites, respectively. The conclusions of these studies were that memantine inhibits and reverses PP-2A inhibition-induced abnormal hyperphosphorylation of tau/neurofibrillary degeneration, and that this drug might be useful for the treatment of AD and related tauopathies.Citation19
Tau and β-amyloid peptides are the main contributors to the pathology of AD. Tau localizes via a 3′-UTR region in its mRNA with an axonic signal sequence.Citation18,Citation19 Because tau is overexpressed, a disturbance in axonal transport could cause hyperphosphorylation events, leading kinesin not to carry out antegrade transport properly,Citation20,Citation21 thus causing a disruption of molecular transport to the axon. Memantine and galantamine produce a reduction in amyloid aggregation,Citation15,Citation17 but show no apparent effect on paired helical filament formation.
Although these drugs increase the availability of the neurotransmitter at the synaptic cleft, the neuronal membrane is damaged so, at best, they may slow progression of symptoms for a few months. This highlights a need to repair the neuronal membranes in order to rehabilitate degenerating neurons to respond better to drug treatments.
Neuronal restoration in AD patients
Why do the drugs available to treat AD have limited efficacy? The answer might be simple. Most of these drugs are acetylcholinesterase inhibitors and allow the neurotransmitter, acetylcholine, to remain for longer in the synaptic cleft. The problem we envisage is that degenerative neurons may not have their cholinergic receptors properly positioned on the damaged membrane, and there may even be some receptors not reaching the postsynaptic neuron during trafficking to the dendrite spine. Restoration of neuronal membranes before starting treatment with an acetylcholinesterase inhibitor might enhance the effects of the drugs to produce a prolonged improvement in symptomatology.
Omega-3 acids
Omega-3 acids have a role in inhibition of ischemic damageCitation22 and also in increased neurite development, demonstrated when hippocampal neurons are treated with docosahexaenoic acid. Omega-3 acids are also involved in the remodeling of membrane raftsCitation23 and in neurogenesis.Citation24 Rafts are microdomains of cell membranes, are rich in cholesterol, and are associated with the stabilization of membrane receptors. Membrane rafts have also been shown to be involved in stabilizing the synapse, the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acidCitation25 and N-methyl-D-aspartate receptors,Citation26 and appear to maintain dendritic spines. Rafts are also required for the proper clustering of acetylcholine receptors.Citation27 When cell membranes are damaged by reactive oxygen species, as occurs in AD, there is a redistribution of lipids, such as cholesterol and sphingomyelin, because of an increase in membrane fluidity,Citation28 and this can lead to changes in the positioning of membrane receptors. Importantly, omega-3 acids have shown the ability to inhibit tau hyperphosphorylation,Citation29 which would allow axonal transport restoration. It has been shown that omega-3 is also capable of reducing amyloid plaque formation.Citation29,Citation30
After neuronal membranes are restored, it would be necessary to re-establish the connections between neurons. This process could be aided by drugs like fluoxetineCitation31 and escitalopram,Citation32 which promote serotonin reuptake. It has been suggested that good recapture of serotonin has a role in the formation of new synapses and neuronal reconnections.
Folic acid
Folic acid has an important role in neuroplasticity and in the maintenance of neuronal integrity. Folate is a cofactor in one-carbon metabolism, during which it promotes the regeneration of methionine from homocysteine, a highly reactive sulfur-containing amino acid. Methionine can then be converted to S-adenosylmethionine, the principal methyl donor in most biosynthetic methylation reactions. At the cellular level, folate deficiency and hyperhomocysteinemia exert multiple detrimental effects. These include induction of DNA damage, misincorporation of uracil into DNA and altered patterns of DNA methylation. Low folate and elevated homocysteine levels increase the generation of reactive oxygen species, and contribute to excitotoxicity and mitochondrial dysfunction, which might lead to apoptosis. Strong epidemiological and experimental evidence links derangements of one-carbon metabolism to vascular, neurodegenerative, and neuropsychiatric disease, in particular cerebral ischemia, Alzheimer’s dementia, and depression.Citation33 Therefore, we believe it would be useful to incorporate treatment with folic acid into neuronal rehabilitation therapy.
Ginkgo biloba
Ginkgo bilboa, a plant extract, has had controversial results in AD patients.Citation34 However, some studies have shown a neuroprotective effect (attributed to its high flavonoid content)Citation35 and an antioxidant action.Citation36 Although it has been suggested that the use of Ginkgo biloba has no benefit in AD patients, there exists some evidence for a beneficial effect from EGb 761 extract. Ginkgo biloba leaves contain a unique kind of flavonoid, and are harvested in South Korea, Japan, and France.Citation37
Studies have demonstrated that β-amyloid peptide treatment induces free radical production and increased glucose uptake, apoptosis, and cell death in PC12 nerve cells. Addition of a standardized extract of Ginkgo biloba leaves, ie, EGb 761, together with β-amyloid peptide prevented β-amyloid peptide-induced free radical production, increased glucose uptake, apoptosis, and cell death, in a dose-dependent manner, However, pretreatment of these nerve cells with EGb 761 did not prevent β-amyloid peptide-induced toxicity, although it did prevent β-amyloid peptide-induced reactive oxygen species generation. Moreover, the terpene and flavonoid-free EGb 761 extract, HE 208, although able to inhibit a β-amyloid peptide-induced increase in glucose uptake, failed to protect the cells against β-amyloid peptide-induced apoptosis and cytotoxicity. These results indicate that the terpenoid and flavonoid constituents of EGb 761, probably acting in combination with other components present in HE 208, are responsible for rescuing neuronal cells from β-amyloid peptide-induced apoptosis and cell death, their mechanism of action being distinct from their antioxidant properties.
Because pre- and post-treatment with EGb 761 does not protect cell-induced neurotoxicity, the possibility that EGb 761 interacts directly with β-amyloid peptide was investigated. Indeed, in vitro reconstitution studies demonstrated that EGb 761 inhibits, in a dose-dependent manner, the formation of β-amyloid peptide-derived diffusible neurotoxic soluble ligands, which are thought to be involved in the pathogenesis of AD.Citation38 Importantly, Ginkgo biloba acts as a protective agent against the formation of amyloid fibrils, and this effect involves the MAP kinase cascade, SIRT1, and beta NFκ.Citation39
Resveratrol
Phytophenols can be arbitrarily divided into the single-ring phenolic acids, the bisphenols, which include stilbenes, tricyclic phenols (flavonoids), and their subclasses, and the oligomeric and polymeric species, proanthocyanidins and anthocyanidins. Phytophenol precursors and stilbenes, including resveratrol and its analogs and conjugates, appear to have a preventive and possibly therapeutic role in atherosclerosis and certain neoplastic and inflammatory conditions. These molecules probably act as free radical scavengers, and might selectively interfere with multiple factors affecting cell division in rapidly and abnormally proliferative mammalian cells. There have been a number of studies published on the natural occurrence, extraction methods, bioavailability, analytical detection, and metabolism of resveratrol, as well as its effects on cancer, the inflammation process, atherosclerosis, and neurons.
Grape extracts are a convenient source of salutary phytochemicals to supplement currently available occidental food. Resveratrol could be added in biosignificant amounts to these extracts if extraction, content, and quality control issues are instituted.Citation40
Resveratrol and catechin have been shown to be protective against β-amyloid peptide in PC12 cells. Many environmental factors, including antioxidants, metal ions, and proteoglycans can modify β-amyloid peptide (1–41) toxicity in PC12 cells. Protection against reactive oxygen species toxicity is concentration-dependent for both resveratrol and catechin. This protective effect appears to be merely additive, rather than synergistic, and is unlikely to be due to antioxidant activity alone. The difference in chemical and biological activity shown by these compounds for several cell types, as well as the complex toxicity of β-amyloid peptide (1–41), may explain this synergistic protective effect. Utilization of different compounds with synergistic activity may improve the protective effect against complex toxicity mechanisms.Citation41 A polyphenol-rich diet could help to maintain brain homeostasis, prevent oxidation, and keep neurons healthy.Citation42
Nimesulide
Inflammatory processes in the brain are at the basis of the pathogenesis of AD, so nonsteroidal anti-inflammatory drugs have a protective effect in affected individuals. In a study in which quisqualic acid was injected into the right nucleus basalis of rats, excitotoxin induced cholinergic degeneration, an intense glial reaction, and production of inflammatory mediators. Seven days of treatment with intramuscular nimesulide 10 mg/kg/day strongly attenuated the microglial reaction, reduced the number of inducible nitric oxide synthase-positive cells, and completely abolished increased prostaglandin E2 formation. These data indicate that cyclo-oxygenase-2 (COX-2) inhibitors have potential efficacy in the treatment of AD.Citation43
COX-2 is involved in the inflammatory component of the ischemic cascade, playing an important role in delaying progression of brain damage. In one study, the neuroprotective effect of nimesulide was still evident 30 days after an ischemic episode, providing the first experimental evidence that COX-2 inhibitors confer long-lasting neuroprotection. Oral administration of nimesulide was also able to reduce the extent of brain damage significantly, which suggests that its protective effects are independent of the route of administration. This study confirms the ability of COX-2 inhibitors to reduce brain damage induced by cerebral ischemia, and indicates nimesulide can provide neuroprotection when administered for up to 24 hours after an ischemic event.Citation44
Nimesulide has been withdrawn from the market in a number of countries because of its side effects, in particular the risk of hepatotoxicity at therapeutic doses. No English-speaking country has approved its use, so there are limited data from Phase IV studies. There have been two reports of death in children as a result of Reye’s syndrome, a condition commonly associated with aspirin consumption in children under 12 years of age, and which causes numerous detrimental effects, especially to the brain and liver. In these cases, Reye’s syndrome was supposedly caused by consumption of nimesulide. In addition, nimesulide was observed to have a dangerously rapid antipyretic effect in children, and is not recommended for reduction of fever under 40°C (approximately 101°F). However, physicians often succumb to parental pressure to lower their child’s fever. In countries like Finland, Spain, and Turkey, use of nimesulide has been prohibited in both children and adults, because of its hepatotoxicity, excessive antipyretic effect, and the potential to cause Reye’s syndrome. The European Drug Evaluation Agency is currently evaluating the status of nimesulide for an all-European approach.
Despite the adverse effects associated with nimesulide, it is beneficial if only given for a limited period of time and, like other nonsteroidal anti-inflammatory drugs, should not be used in children.
Recommendations
To date, we have followed two AD patients trying this therapy. Both patients were on drug treatment but showing no improvement. In addition to their existing treatment, they started our proposed therapy approximately one year ago, with satisfactory results so far.
Case 1
An 83-year-old woman was diagnosed with AD about 10 years ago. At the time the proposed therapy was initiated, the patient presented with confusion about money, words, and the placing of objects, in addition to difficulty in remembering certain activities and even recognizing her relatives. She had been taking rivastigmine, but after two years she started showing marked cognitive decline. After one year taking our proposed therapy, she now shows an improvement in memory, fluidity of language, and recognizes her relatives again. One of her sons said that the disease seems to have stopped, and that her brain appears to be regaining functionality.
Case 2
A man with a dramatic picture of AD had presented with very rapid cognitive decline, despite being treated with memantine. His wife made the decision to start this therapy, and his memory and conversational coherence has improved markedly. The only residual sign of the disease in this man is occasional mild aggression.
Discussion
Omega-3 acid could help restoration of neuronal membranes to the AD brain, allowing proper positioning of postsynaptic receptors, and enabling existing drug treatment to be more effective. Tau dephosphorylation would restore axonal transport of synaptic vesicles, and thus improve neurotransmitter release at the axon terminals. Once the membranes have been repaired, it would be important to induce the formation and maintenance of new synapses () by fluoxetine or escitalopram and folic acid. Serotonin regulates neuronal morphology and the reconnection between neurons,Citation45 its pathway interacts with that of acetylcholine,Citation46 and it also has a role in memory impairment.Citation47,Citation48 A powerful antioxidant, such as resveratrol, and the activation of memory processes induced by Ginkgo bilboa, would improve the effects of membrane repair and formation of synapses. All of this together might then rehabilitate the AD brain to enable institution of drug treatment producing a better outcome (). Ongoing neurodegenerative processes and the mechanisms of action of the drugs available have failed to cure AD,Citation49 because they are brain enhancers.Citation50
Figure 1 Restoration of the dendritic spine. Dendrites damaged as a result of Alzheimer’s disease would result in abnormal neurotransmission because the receptors are not located in the right site as a result of the effect of neurodegeneration. The repair process is shown, beginning with omega-3 to allow repair of membranes and thus locate the receptors in the right places for effective neurotransmission. Serotonergic neurons would help in the formation of circuits due to the use of serotonin reuptake. Resveratrol and Ginkgo biloba serve as antioxidants and in the process of memory, respectively. Folic acid maintains the integrity of the newly repaired circuits.
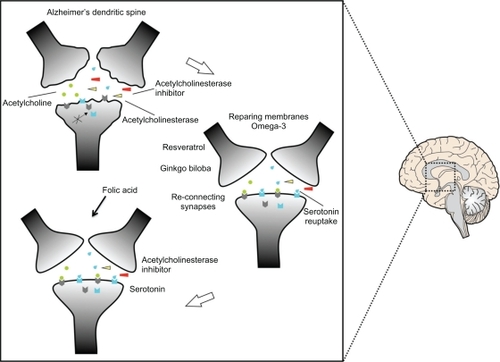
Figure 2 Alternative therapy to rehabilitate a neuron.

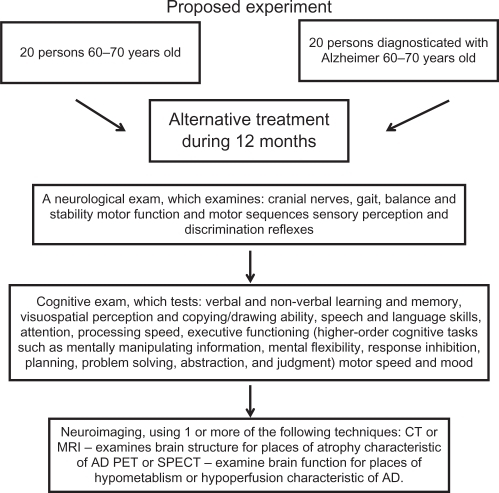
We believe this rehabilitating therapy in combination with drug treatment gives us a good possibility for improving the quality of life of both AD patients and their relatives, who also suffer the consequences of the disease. We propose an experimental strategy to evaluate the effects of this therapy (). Of course, this should be tested in more AD patients in the near future.
Figure 3 Proposed experiment to probe the alternative therapy.
We must bear in mind that AD has a slow progression and the neuronal damage occurs over a long period of time, to such an extent that it becomes impossible to prevent brain cell degeneration. At the last congress of The Society of Neuroscience at Chicago in 2009, the introduction of natural products to treat AD was recommended.
The alternative therapy we propose here could take over a year to have an effect, but it might be really helpful in patients with mild to moderate AD. However, in the advanced stages of the disease, this therapy would not be appropriate due to the large amount of neuronal loss. Although this therapy requires patience and cooperation from both patients and their relatives, we believe it provides a good option to improve treatment outcomes in AD.
Disclosure
The authors report no conflicts of interest in this work.
References
- Perez-TurJGenetics and Alzheimer’s diseaseRev Neurol20003016116910730324
- Lopez de MunainAClassification of mitochondrial diseasesRev Neurol199826Suppl 1S9S149810585
- HardyJAllsopDAmyloid deposition as the central event in the aetiology of Alzheimer’s diseaseTrends Pharmacol Sci1991123833881763432
- JarrettJTLansburyPTJSeeding “one-dimensional crystallization” of amyloid: A pathogenic mechanism in Alzheimer’s disease and scrapieCell199373105510588513491
- HuXHicksCWHeWBACE1 modulates myelination in the central and peripheral nervous systemNat Neurosci200691520152517099708
- WillemMLammichSHaassCFunction, regulation and therapeutic properties of beta-secretase (BACE1)Semin Cell Dev Biol20092017518219429494
- NguyenVHawkinsCBergeronCLoss of nicastrin elicits an apoptotic phenotype in mouse embryosBrain Res20061086768416626651
- GauthierSScheltensPCan we do better in developing new drugs for Alzheimer’s diseaseAlzheimers Dement2009548949119896588
- QizilbashNBirksJLopez ArrietaJLewingtonSSzetoSWITHDRAWN: Tacrine for Alzheimer’s diseaseCochrane Database Syst Rev2007CD00020217636619
- AlfirevicAMillsTCarrDTacrine-induced liver damage: An analysis of 19 candidate genesPharmacogenet Genomics2007171091110018004213
- TsunoNDonepezil in the treatment of patients with Alzheimer’s diseaseExpert Rev Neurother2009959159819402770
- WinbladBDonepezil in severe Alzheimer’s diseaseAm J Alzheimers Dis Other Demen20092418519219246572
- BirksJGrimley EvansJIakovidouVTsolakiMHoltFERivastigmine for Alzheimer’s diseaseCochrane Database Syst Rev2009CD00119119370562
- AnnicchiaricoRFedericiAPettenatiCCaltagironeCRivastigmine in Alzheimer’s disease: Cognitive function and quality of lifeTher Clin Risk Manag200731113112318516265
- MatharuBGibsonGParsonsRGalantamine inhibits beta-amyloid aggregation and cytotoxicityJ Neurol Sci2009280495819249060
- MeloJBSousaCGarcaoPOliveiraCRAgostinhoPGalantamine protects against oxidative stress induced by amyloid-beta peptide in cortical neuronsEur J Neurosci20092945546419222556
- Martinez-CoriaHGreenKNBillingsLMMemantine improves cognition and reduces Alzheimer’s-like neuropathology in transgenic miceAm J Pathol201017687088020042680
- Aranda-AbreuGEBeharLChungSFurneauxHGinzburgIEmbryonic lethal abnormal vision-like RNA-binding proteins regulate neurite outgrowth and tau expression in PC12 cellsJ Neurosci1999196907691710436048
- AronovSArandaGBeharLGinzburgIAxonal tau mRNA localization coincides with tau protein in living neuronal cells and depends on axonal targeting signalJ Neurosci2001216577658711517247
- DubeyMChaudhuryPKabiruHSheaTBTau inhibits anterograde axonal transport and perturbs stability in growing axonal neurites in part by displacing kinesin cargo: Neurofilaments attenuate tau-mediated neurite instabilityCell Motil Cytoskeleton200865899918000878
- LippensGSillenALandrieuITau aggregation in Alzheimer’s disease: What role for phosphorylationPrion20071212519164903
- ReltonJKStrijbosPJCooperALRothwellNJDietary N-3 fatty acids inhibit ischaemic and excitotoxic brain damage in the ratBrain Res Bull1993322232268374800
- FanYYMcMurrayDNLyLHChapkinRSDietary (n-3) polyunsaturated fatty acids remodel mouse T-cell lipid raftsJ Nutr20031331913192012771339
- BeltzBSTlustyMFBentonJLSandemanDCOmega-3 fatty acids upregulate adult neurogenesisNeurosci Lett200741515415817240063
- HeringHLinCCShengMLipid rafts in the maintenance of synapses, dendritic spines, and surface AMPA receptor stabilityJ Neurosci2003233262327112716933
- HouQHuangYAmatoSSnyderSHHuganirRLManHYRegulation of AMPA receptor localization in lipid raftsMol Cell Neurosci20083821322318411055
- Stetzkowski-MardenFRecouvreurMCamusGCartaudAMarchandSCartaudJRafts are required for acetylcholine receptor clusteringJ Mol Neurosci200630373817192619
- ClementABGimplGBehlCOxidative stress resistance in hippocampal cells is associated with altered membrane fluidity and enhanced nonamyloidogenic cleavage of endogenous amyloid precursor proteinFree Radic Biol Med2010481236124120156550
- MaQLYangFRosarioERBeta-amyloid oligomers induce phosphorylation of tau and inactivation of insulin receptor substrate via c-Jun N-terminal kinase signaling: suppression by omega-3 fatty acids and curcuminJ Neurosci2009299078908919605645
- AmtulZUhrigMRozmahelRFBeyreutherKStructural basis for the differential effects of omega-3 and omega-6 fatty acids on Abeta production and amyloid plaquesJ Biol Chem20101022 [Epub ahead of print]
- WangJWDavidDJMoncktonJEBattagliaFHenRChronic fluoxetine stimulates maturation and synaptic plasticity of adult-born hippocampal granule cellsJ Neurosci2008281374138418256257
- LucasGDuJRomeasTSelective serotonin reuptake inhibitors potentiate the rapid antidepressant-like effects of serotonin4 receptor agonists in the ratPLoS One20105e925320169084
- KronenbergGCollaMEndresMFolic acid, neurodegenerative and neuropsychiatric diseaseCurr Mol Med2009931532319355913
- OkenBSStorzbachDMKayeJAThe efficacy of Ginkgo biloba on cognitive function in Alzheimer’s diseaseArch Neurol199855140914159823823
- BastianettoSZhengWHQuirionRThe Ginkgo biloba extract (EGb 761) protects and rescues hippocampal cells against nitric oxide-induced toxicity: Involvement of its flavonoid constituents and protein kinase CJ Neurochem2000742268227710820186
- GrundmanMGrundmanMDelaneyPAntioxidant strategies for Alzheimer’s diseaseProc Nutr Soc20026119120212133201
- DrieuKPreparation and definition of Ginkgo biloba extractPresse Med198615145514572947081
- YaoZDrieuKPapadopoulosVThe Ginkgo biloba extract EGb 761 rescues the PC12 neuronal cells from beta-amyloid-induced cell death by inhibiting the formation of beta-amyloid-derived diffusible neurotoxic ligandsBrain Res200188918119011166702
- LongpreFGarneauPChristenYRamassamyCProtection by EGb 761 against beta-amyloid-induced neurotoxicity: Involvement of NF-kappaB, SIRT1, and MAPKs pathways and inhibition of amyloid fibril formationFree Radic Biol Med2006411781179417157181
- SovakMGrape extract, resveratrol, and its analogs: A reviewJ Med Food200149310512639418
- ConteAPellegriniSTagliazucchiDSynergistic protection of PC12 cells from beta-amyloid toxicity by resveratrol and catechinBrain Res Bull200362293814596889
- RossiLMazzitelliSArcielloMCapoCRRotilioGBenefits from dietary polyphenols for brain aging and Alzheimer’s diseaseNeurochem Res2008332390240018415677
- ScaliCProsperiCVannucchiMGPepeuGCasamentiFBrain inflammatory reaction in an animal model of neuronal degeneration and its modulation by an anti-inflammatory drug: Implication in Alzheimer’s diseaseEur J Neurosci2000121900191210886331
- Candelario-JalilEAlvarezDGonzalez-FalconANeuroprotective efficacy of nimesulide against hippocampal neuronal damage following transient forebrain ischemiaEur J Pharmacol200245318919512398903
- DaubertEACondronBGSerotonin: A regulator of neuronal morphology and circuitryTrends Neurosci20103342443420561690
- AltmanHJStoneWSOgrenSOEvidence for a possible functional interaction between serotonergic and cholinergic mechanisms in memory retrievalBehav Neural Biol19874849623632552
- BuhotMCMartinSSeguLRole of serotonin in memory impairmentAnn Med20003221022110821328
- DoughertyJJNicholsRACross-regulation between colocalized nicotinic acetylcholine and 5-HT3 serotonin receptors on presynaptic nerve terminalsActa Pharmacol Sin20093078879419498419
- National Institutes of HealthAlzheimer′s Disease Medications2008 Available from: http://www.nia.nih.gov/Alzheimers/Publications/medicationsfs.htm. Accessed on January 18, 2010
- StixGTurbocharging the brainSci Am2009301464919780452
- JhaAMAbha. Assessment of cytotoxic and clastogenic effects of nimesulide: An NSAID drug in somatic cells of BALB/c mice in vivoDrug Chem Toxicol20103320420820307146
- GoncalvesMBWilliamsEJYipPYanez-MunozRJWilliamsGDohertyPThe COX-2 inhibitors, meloxicam and nimesulide, suppress neurogenesis in the adult mouse brainBr J Pharmacol20101591118112520136845