Abstract
Hutchinson–Gilford Progeria Syndrome and Werner syndrome, also known as childhood- and adulthood-progeria, respectively, represent two of the best characterized human progeroid diseases with clinical features mimicking physiological aging at an early age. The discovery of their genetic basis has led to the identification of several gene mutations leading to a spectrum of progeroid phenotypes ranging from moderate and mild–severe to very aggressive forms. In parallel, the creation of disease registers and databases provided available data for the design of relatively large-scale epidemiological studies, thereby allowing a better understanding of the nature and frequency of the premature aging-associated signs and symptoms. The aim of this article is to review the most recent findings concerning the epidemiology of premature aging disorders, their genetic basis, and the most recent reports on the frequency of associated diseases.
Introduction
Hutchinson–Gilford Progeria Syndrome (HGPS) and Werner syndrome (WS) are two of the best characterized human progeroid diseases with clinical features mimicking physiological aging at an early age, the first being referred as to childhood progeria, and the latter as progeria of adulthood.Citation1 An increasing number of intermediate progeroid phenotypes, known as atypical progeroid syndromes, atypical HGPS, or atypical WS, have been described.Citation1 The aim of this review article is to describe the most recent findings concerning the epidemiology of premature aging disorders, their genetic basis, and the most recent reports on the frequency of associated diseases.
Hutchinson–Gilford Progeria Syndrome
HGPS is an extremely rare genetic disorder affecting about one per four to eight million live births.Citation2 More precisely, the reported prevalence rate of the disease is one in eight million births, but if unreported or misdiagnosed cases are taken into account, the estimated birth prevalence is one in four million.Citation2
According to the Progeria Research Foundation database (http://www.progeriaresearch.org/prf-by-the-numbersprf.html), there are an estimated 200–250 children living with progeria worldwide at any one time, and 103 of them have been identified as of April 2013. Progeria affects both sexes and all races, and HGPS cases have been discovered in over 40 different countries. In particular, a map of ascertained HGPS children is available at the Progeria Research Foundation portal (http://www.fidtheother150.org/), and there are records of 20 cases in Northern America, 16 cases in Central and Southern America, 24 cases in Europe and the Mediterranean regions, four cases in Africa, and 18 cases in Asia.
The disease was named after the reports by Jonathan Hutchinson and Hastings Gilford, the doctors who first described it in England, and it is classified as a segmental progeroid syndrome since multiple organs and tissues replicate phenotypes associated with normal aging.Citation3,Citation4
Children with HGPS appear healthy at birth but develop distinctive clinical features during the first years of their life, including severe growth retardation, usually associated with skeletal alteration as well as loss of subcutaneous fat and skin appendages, and some developmental processes are delayed (dentition) or absent.Citation4 Death occurs in those affected by their early teenage years, and usually results from heart attacks and strokes.Citation1
The majority of classical HGPS is caused by a de novo point mutation in exon 11 of the LMNA gene (c.1824C>T, p.G608G).Citation5 The LMNA gene encodes A-type lamins, which are intermediate filament proteins of the inner nuclear lamina. The c.1824C>T mutation results in the activation of a cryptic splice donor site that removes 150 nucleotides from exon 11. The resulting lamin A∆150 messenger ribonucleic acid gives rise to a lamin A isoform containing an internal deletion of 50 amino acids, known as progerin (a protein that cannot undergo complete maturation).Citation5 Lamins constitute the major component of the nuclear lamina; in addition to providing structure and shape to the nucleus, they are involved in organizing several processes including chromatin organization, deoxyribonucleic acid (DNA) replication, transcription, DNA methylation and epigenetic regulation, and DNA repair.Citation6 Those mechanisms are impaired in HGPS and might contribute to the progeria phenotype. Indeed, cells obtained from HGPS patients show a markedly reduced lifespan when grown in culture, and accumulate defects in nuclear structure and architecture with cell passaging, including lobulation of the nuclear envelope, thickening of the nuclear lamina, loss of peripheral heterochromatin, and clustering of nuclear pores, which are accompanied by an increase in the amount of progerin within the cells.Citation7
HGPS-associated symptoms
A visible vein across the nasal bridge is often the first observable sign in HGPS infants. A profound failure to thrive occurs during the first year, usually from 6 months to 12 months. On average, HGPS children gain 0.4–0.5 kg/year and reach a final weight of about 14–15 kg and a final height of 110 cm. Progressive alopecia usually takes place within 6 months to 2 years, and between the ages of 2 years and 3 years, most children become bald.Citation8 Other symptoms become apparent during the first year to third year, including characteristic facies, loss of subcutaneous fat, stiffness of joints, bone changes, and abnormal tightness of the skin over the abdomen and upper thighs.Citation1 With time, the skin becomes thin, dry, and atrophic, with reduced turgor and sometimes with hyperkeratosis. Small, light-brown spots frequently develop on the neck and upper thorax, and subsequently on the scalp and limbs. Typical facial abnormalities include a receding mandible, a small and beaked nose, prominent scalp veins, prominent eyes, and protruding ears that lack lobules. The facial characteristics gradually develop and both the face and body change with time: the subcutaneous fat in the face disappears completely and the facial muscles decrease in size. The body shows increasing loss of subcutaneous fat and muscle bulk and the joints protrude.Citation1,Citation8,Citation9 At the bone level, patients show clavicular hypoplasia, generalized osteopenia, and acroosteolyses of distal phalanges. Motor and mental development is normal, cognitive functions are preserved, and the children follow a normal psychosocial development and show normal behaviors for their age.Citation1,Citation9 Dentition is delayed and crowded.Citation9 HGPS individuals have a high-pitched voice, do not reproduce, and their appearance becomes like that of an older person with time.Citation1,Citation8
Additional findings that are present in some but not all affected individuals include excessive ocular tearing, photophobia, exposure keratitis, and Raynaud’s phenomenon.Citation9 Most children die in their early teens from heart attacks and strokes caused by progressive atherosclerotic disease, with myocardial infarction representing the most frequent cause of death at a mean age of around 13 years.Citation8 A comprehensive description of most of the HGPS-associated manifestations is provided in the following sections.
Cardiovascular disease in HGPS
Cardiovascular disease (CVD) represents the principal factor affecting mortality in HGPS individuals, with death resulting from myocardial infarction, stroke, or congestive cardiac failure in 75% of cases.Citation10 Cardiovascular problems are absent during the first 5 years of life, but children gradually develop shortness of breath with exertion and easy fatigability from the age of 6–8 years.Citation10 Accelerated CVD leads to debilitating morbidity in HGPS and culminates in mortality from myocardial infarction or stroke at an average age of 13 years.Citation11
The rapid progression of CVD in HGPS presents an opportunity to explore the natural history of human CVD, and a study performed on 26 HGPS patients and 22 matched controls revealed that the carotid–femoral pulse wave velocity was dramatically elevated in patients.Citation11 Carotid duplex ultrasound echobrightness, assessed as a measure of arterial wall density, was significantly greater than age- and sex-matched controls in the intima–media, near adventitia, and deep adventitia, as was internal carotid artery mean flow velocity.Citation11 Overall, those data demonstrated that, along with peripheral vascular occlusive disease, accelerated vascular stiffening is an early and pervasive mechanism of vascular disease in HGPS.Citation11
Autopsy data have shown widespread atherosclerosis in HGPS patients. Particularly, advanced coronary atherosclerotic lesions have been reported, and the arteries were frequently stenosed or occluded by plaques or narrowing of intramural arteries. Occlusion of the right coronary artery, lesions of the left anterior descending artery, and severe atherosclerosis of the aorta, represent common findings in HGPS.Citation10 Valvular changes and pulmonary arterial lesions have also been reported in HGPS individuals.Citation10
Cerebrovascular disease in HGPS
Cerebrovascular arteriopathy and stroke have been recently assessed by means of a neurovascular imaging cohort study of HGPS, a study aimed to identify the neurovascular features, infarct type, topography, and natural history of stroke.Citation12 A total of 25 children with confirmed diagnoses of HGPS were included in the study, which revealed a vasculopathy unique to HGPS, including distinctive intracranial stenoocclusive arterial lesions, basal cistern collateral vessels, and slow compensatory collateral flow over the cerebral convexities. Moreover, the authors identified early and clinically silent strokes as a prevalent disease characteristic in HGPS. Indeed, a radiographic evidence of infarction was found in 60% of patients, of which half were likely clinically silent.Citation12
Skeletal abnormalities in HGPS
A recent study performed with an inducible and tissue-specific mouse model, which expresses the most common HGPS mutation (c.1824C>T) in osteoblasts and odontoblasts, revealed that the expression of the HGPS mutation during osteoblast development results in a loss of osteocytes, irregular mineralization, and poor biomechanical properties.Citation13
A comprehensive survey of the skeletal dysmorphisms observed in children with HGPS using conventional radiography was obtained from 39 children with the classic HGPS genotype, representing approximately 15%–20% of the world’s HGPS population.Citation14 Small clavicles were observed in 100% of the patients; followed by coxa valga and acroosteolysis, which were observed in more than 90% of the patients; and resorption of the distal clavicles and narrow apices, both present in 82% of the subjects. Other frequent skeletal abnormalities were hip dysplasia (69%) and thin ribs (59%). In addition, 30% to 45% of the patients showed resorption of the anterior ribs, closed sagittal suture, generalized osteopenia, focal cortical defects, flexed fingers, ulnar minus variant, or enlarged heart. Less frequent (20%–30%) were dystrophic calcification, sagittal suture diastasis, enlarged femoral head, pseudoarthrosis, enlarged femoral greater trochanter, and avascular necrosis of the proximal femur. Kyphoscoliosis, enlarged humoral head, narrowed humoral diaphysis, prominent pulmonary vessels, and wormian bones were reported in less than 15% of the patients, and other abnormalities, such as accentuated osteopenia of proximal humoral/femoral epiphysis, rib fracture, Madelung deformity, ivory epiphyses, bifid rib, and congenitally fused ribs were reported in 10% or less of the patients.Citation14
Craniofacial abnormalities in HGPS
Using the Progeria Research Foundation medical and research database (http://www.progeriaresearch.org/medical_database.html), data on 25 HGPS patients have been examined in order to provide an overview of the craniofacial abnormalities in progeria.Citation15
Concerning scalp, calvarial, and skull base features, thinning of the calvarium was seen in 95% of the individuals, often accompanied by a paucity of scalp fat (91%). A mottled appearance of the skull was seen in 59% of the patients. Two individuals (8%) had skull fractures, and prominent vascular markings of the bony calvaria were observed in 90% of the subjects. Craniofacial disproportion (a large cranium relative to the facial size), and a J-shaped sella were observed in almost 90% of the patients, and a delayed closure of the anterior fontanel was seen in 56% of them.Citation15
Concerning oral maxillary, zygomatic arch, and parotid gland features, the authors observed a short mandibular ramus in 83% of the patients, with a gracile thin zygomatic arch in 50% of them. A shallow glenoid fossa with a hypoplastic or absent articular eminence and flattening of the mandibular condyle were seen in 43% of the patients. Moreover, 45% of the children had a V-shaped palate, and 50% of them had disorganized dentition. A prominent parotid gland was seen in all the children analyzed.Citation15
With regard to orbital features, hypotelorism was noted in 86% of the children, and kinking of the optic nerves was seen in 89%.Citation15
Other manifestations in HGPS
Fifteen patients with HGPS have been enrolled in a prospective study to evaluate otologic and audiologic manifestations.Citation16 All patients had stiff auricular cartilages, small or absent lobules, and hypoplasia of the lateral soft-tissue portion of the external ear canal leading to a shortened canal. A low-frequency conductive hearing loss in the 250 Hz to 500 Hz range was observed in 86.4% of the ears, despite largely normal tympanometry.Citation16 In addition, 71% of the patients had dry cerumen impaction, and 29% of them reported a history of recurrent otitis media.Citation16
Insulin resistance occurs in about 50% of affected patients without progression to diabetes mellitus.Citation9 A rare case of a 10-year-old boy with genetically confirmed classical HGPS and hypoparathyroidism has been reported.Citation17 Differently from other premature aging syndromes, such as WS or others caused by mutations of DNA repair genes, there is no reported increase of cancer incidence in HGPS patients.Citation8,Citation18 Cataracts are not frequent.Citation8 We recently reviewed neurodegenerative signs or symptoms in premature aging disorders, and they are absent in HGPS individuals.Citation1
From mild to severe progeria: the genetic basis of classical HGPS and atypical progeria syndromes
Although the majority (approximately 90%) of classical HGPS is caused by a de novo point mutation in exon 11 of the LMNA gene (c.1824C>T, p.G608G), it was clear from the beginning that other mutations in LMNA could cause a similar phenotype.Citation5 Particularly, the genetic basis of HGPS was identified in 2003 by two independent research groups.Citation5,Citation19 Eriksson et alCitation5 observed the G608G mutation in 90% of 20 HGPS individuals, and De Sandre- Giovannoli et alCitation19 observed it in two affected children. A patient with a c.1822G>A mutation (p.G608S), with classical HGPS, was also recorded by Eriksson et al.Citation5
The LMNA gene encodes the four different A-type lamins (lamin A, lamin A∆10, lamin C, and lamin C2), which are intermediate filament proteins of the inner nuclear lamina.Citation1 Lamin A (encoded by exons 1–12) and lamin C (encoded by exons 1–10) are the major proteins expressed in differentiated cells. Lamin A∆10 is identical to lamin A except that it lacks exon 10 and has been detected in cells from colon and placenta, in leukocytes and fibroblasts, and in tumor cells. Lamin C2 has an alternative exon 1 compared with lamin C and is present in germ cells.Citation1 Lamin A proteins contain CaaX boxes at their C-terminal ends; they are synthesized as prelamin A proteins, which undergo farnesylation and other posttranslational modifications to become mature proteins ().
Figure 1 Representation of prelamin A processing and summary of LMNA and ZMPSTE24 mutations leading to classical and atypical progeria.
Abbreviations: HGPS, Hutchinson–Gilford Progeria Syndrome; ZMPSTE24, zinc metalloprotease related to Ste24p; FTASE, farnesyltransferase; RCE1, Ras-converting enzyme 1; ICMT, isoprenylcysteine carboxyl methyltransferase; WS, Werner syndrome.
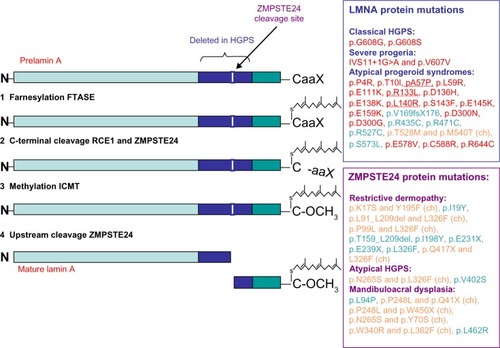
The p.G608G mutation results in the activation of a cryptic splice donor site, leading to the production of a lamin A isoform containing an internal deletion of 50 amino acids, known as progerin – a protein that cannot undergo complete maturation.Citation5 Particularly, the deletion eliminates the site for endoproteolitic cleavage by zinc metalloproteinase Ste24 homolog (ZMPSTE24), a cleavage required for the production of mature lamin A. The recurrent c.1824C>T mutation causing HGPS is a de novo dominant point mutation, mostly originating on the paternal allele and often linked with advanced paternal age.Citation5
The zinc metalloprotease, ZMPSTE24, plays a critical role in nuclear lamin biology by cleaving the prenylated and carboxylmethylated 15-amino acid tail from the C-terminus of prelamin A to yield mature lamin A (). Two patients with extraordinarily severe forms of progeria caused by unusual mutations in LMNA have been described.Citation20 Both mutations (IVS11+1G>A and p.V607V) resulted in a strong activation of the aberrant splice site observed in typical HGPS, leading to increased progerin expression compared to typical HGPS cases.Citation20 By contrast, two subjects bearing a missense (p.T623S) mutation leading to the deletion of 35 amino acids in exon 11 of LMNA showed a less aggressive progeroid phenotype (slowly progressing progeria) compared to classical HGPS.Citation21,Citation22 Overall, the amount of toxic progerin in cells appears to correlate with the severity of disease outcomes.Citation20
Interestingly, recessive mutations in ZMPSTE24 also disrupt lamin A proteolytic processing and are associated with three distinct but related human diseases that share features of premature aging, with a gradation of severity.Citation23 Indeed, ZMPSTE24 mutations cause: (1) the mild progeroid disorder mandibuloacral dysplasia, a rare autosomal recessive disorder characterized by postnatal growth retardation, craniofacial anomalies, skeletal malformations, and mottled cutaneous pigmentation; (2) a severe form of progeria denoted as atypical HGPS; and (3) restrictive dermopathy (RD), a fatal neonatal disorder characterized by severe intrauterine growth delay that can be considered as an “extreme form” of premature aging.Citation23 Twenty human ZMPSTE24 alleles have been identified that are associated with those diseases. They have been recently reviewed by Barrowman et al,Citation24 who demonstrated a correlation between decreasing ZMPSTE24 protease activity and increasing disease severity. Particularly, complete loss-of-function alleles are associated with RD, whereas retention of partial, measurable activity results in mandibuloacral dysplasia or severe progeria.Citation23 illustrates most of the known LMNA and ZMPSTE24 mutations linked to progeroid diseases.
Although the mechanism whereby persistently farnesylated lamin A, either resulting from LMNA or ZMPSTE24 mutations, causes premature aging phenotypes is unknown, changes in chromatin architecture and in histone methylation and gene expression, defective DNA repair and accumulation of DNA damage, impaired structural and mechanical properties of the nuclear lamina, and perturbations in transcription factors and nuclear proteins, are among the suggested pathologic mechanisms.Citation1,Citation8
The diagnosis of HGPS is based on the recognition of common clinical features and the detection of either the c.1824C>T (p.G608G) heterozygous LMNA mutation in the classic form of HGPS, or one of three of the heterozygous LMNA mutations in atypical HGPS: c.1822G>A (p.G608S), c.1821G>A (p.V607V), or c.1968+1G>A (IVS11+1G>A) ().Citation9 Noteworthy, several patients with atypical progeroid syndromes, which have been referred to as atypical progeroid syndromes (also called atypical HGPS, nonclassical progeria, or atypical WS), do not carry LMNA splicing mutations, but other heterozygous, homozygous, or compound heterozygous mutations in the LMNA gene.Citation24 The clinical features of those patients include growth retardation and involve the same body systems (bones, body fat, skin, and hair) as in classical HGPS, but the course and severity of the symptoms vary. The patients have differing ages of onset and symptom severity, with some nearly as severe as HGPS, others (such as p.R435C) leading to atypically mild RD, but most are far less severe.Citation24 For example, patients affected by atypical WS have early onset of aging phenotypes and an accelerated rate of disease progression than typical WS individuals; they also commonly show absence of bilateral cataracts and diabetes, which are common features in typical WS.Citation25 Over 20 LMNA mutations causing atypical progeroid syndromes have been discovered and have been recently reviewed by Doubaj et al;Citation23 these are summarized in .
In this regard, a dominantly inherited premature aging syndrome that includes prominent cardiovascular and cutaneous manifestations, called LMNA-associated cardiocutaneous progeria syndrome, was recently described.Citation26 The disease showed a later onset than classical HGPS, with skin manifestations of aging appearing in the third decade of life. Affected individuals also developed CVD leading to death in their fourth decade. One affected individual in this family also showed primary malignancies not previously reported in LMNA-linked progeria disorders. This atypical progeroid syndrome was caused by a novel LMNA mutation (c.899A>G, p.D300G) leading to abnormalities of the nuclear membrane architecture.Citation26
There is also indication from studies in nonagenarians suggesting that common LMNA polymorphisms and haplotypes may play a role in the human lifespan.Citation27 In this regard, Rodríguez and ErikssonCitation28 have recently observed that an LMNA single nucleotide polymorphism (rs4641) results in low and high expressing alleles of the LMNA gene, and might account for the variability in phenotype seen among HGPS individuals.Citation28 Noteworthy, not all LMNA mutations cause progeria. More than 450 mutations of the gene have been described causing several different autosomal dominant or recessive diseases collectively called primary laminopathies, and these include muscular dystrophies, lipodystrophies, neuropathies, cardiomyopathies, and the above-discussed segmental progeroid syndromes.Citation26,Citation29
Werner syndrome
WS is a rare autosomal recessive disorder also called adult progeria, and represents the most studied disease model of premature aging in adulthood.Citation30 In the Japanese population, a founder effect has been described, and the frequency of WS has been roughly estimated to be 1:100,000.Citation30 Another cluster of WS has been identified in Sardinia, with 18 described cases due to a founder effect.Citation31 The prevalence of heterozygous carriers in Japan is approximately 1/167, and it is estimated to be approximately 1/120 in Sardinia.Citation31 Outside of Japan the disease prevalence is estimated to be approximately 1:1,000,000–1:10,000,000.Citation30
The syndrome was first described in the doctoral thesis of WernerCitation32 in 1904. According to a recent report, 1,487 WS cases have been recorded from 1904 to the end of 2008 – 1,128 in Japan and 359 outside Japan.Citation33 The patients develop features reminiscent of premature aging beginning in the second decade of life, including grey hair, alopecia, prematurely aged face with beaked nose, skin atrophy with scleroderma-like lesions, ischemic heart disease, osteoporosis, bilateral cataracts, type 2 diabetes mellitus, lipodystrophy, and hypogonadism. They also experience an increased risk of cancers, and in most cases, they die because of malignant tumors or arteriosclerosis during the fourth and fifth decades of life.Citation30
The disease is caused by mutations of the WRN gene, which encodes the WRN protein, a member of the RecQ DNA helicase family.Citation34 WRN is a multifunctional nuclear protein that maintains genome stability by means of DNA-dependent adenosine triphosphatase (ATPase), 3′→5′ helicase, 3′→5′ exonuclease, and DNA strand annealing activities.Citation35 Most of the WRN mutations result in the production of truncated proteins lacking the nuclear localization signal, with the subsequent absence of functional WRN protein in nuclei.Citation36 WRN has several functional domains and is considered to be a “caretaker of the genome” since it participates in distinct DNA metabolic pathways, including DNA replication, DNA recombination, telomere maintenance, apoptosis, and DNA repair.Citation36 Cells isolated from WS individuals display increased chromosomal aberrations and premature senescence in culture, as well as accelerated telomere shortening and several defects in DNA replication.Citation37
In 20% of cases, WS is not caused by WRN gene mutations, but often by mutations in the LMNA gene.Citation25 As discussed in the previous section of this paper, WS caused by LMNA mutations is referred to as atypical Werner’s syndrome.Citation25
WS-associated symptoms
The lack of a pubertal growth spurt during the teen years is the first clinical sign in WS individuals, leading to a characteristic short stature and low bodyweight. A recent analysis of 196 WS Japanese cases reported that mean height and bodyweight were 158.3 cm and 45.3 kg for male patients, and 148.5 cm and 37.7 kg for female patients, respectively.Citation38 In their 20s and 30s, WS individuals begin to manifest alopecia, greying hair, and scleroderma-like skin changes, followed by bilateral cataracts, type 2 diabetes mellitus, hypogonadism, skin ulcers, and osteoporosis.Citation1
A recent epidemiological study of 196 cases revealed that greying or loss of hair, bird-like faces, cataracts, and skin atrophy were present in 93% to 99%.Citation38 Other common features were clavus or callus, skin ulcers, flat foot, abnormality of the voice, and calcification in the Achilles tendon, observed in 80%–90% of the subjects.Citation38 Similarly, a trend analysis in Japanese WS individuals revealed that bird-like faces and a stocky trunk with extremely thin extremities are still a hallmark of the disease, but the body size, which is also still small, has been expanding in recent years, in concert with the growing constitution of the general Japanese population, and some patients exceeded 177 cm in height and weighed over 70 kg.Citation33
Abnormal glucose and lipid metabolism, hypogonadism, and bone deformity appear by the fourth decade of life in WS subjects.Citation38 Impaired glucose tolerance is reported in 15%–20% of WS subjects, diabetes mellitus in 55%–70%, and dyslipidemia in 60%–85%.Citation38 Fertility in WS patients appears to decline soon after sexual maturity, and hypogonadism has been reported in 40% of both sexes.Citation38 Osteoporosis was observed in more than 60% of WS cases, while osteoarthritis has not been frequently reported in WS.Citation33,Citation38
For several years, the clinical diagnosis of WS was based on the presence of four cardinal signs (cataracts, skin changes, short stature, and greying or loss of hair), which are observable in more than 95% of the cases, as well as on additional signs (osteoporosis, voice change, atherosclerosis, type 2 diabetes mellitus, and so on). For a definite diagnosis, all the cardinal signs (onset over 10 years old) and two additional signs should have been present. In addition, sequencing of the WRN gene could be performed, and the absence of normal WRN protein would be confirmed by Western blot analysis.Citation4,Citation35
Following more recent observations, revised diagnostic criteria have been proposed,Citation38 including the following cardinal signs and symptoms (onset over 10 years until 40 years of age): (1) progeroid changes of the hair (gray hair, baldness, and so on); (2) cataracts (bilateral); (3) changes of the skin (atrophic skin, tight skin, clavus, callus); (4) soft-tissue calcification (Achilles tendon, and so on); (5) bird-like face; and (6) abnormal voice (high-pitched, squeaky, hoarse). Additional signs include abnormal glucose and/or lipid metabolism, deformation and abnormality of the bone (osteoporosis, and so on), malignant tumors, parental consanguinity, premature atherosclerosis, hypogonadism, short stature, and low bodyweight. The diagnosis is confirmed if all the cardinal signs are present, or if there is a gene mutation in addition to at least three cardinal signs, and suspected if two or more cardinal signs are present, or if 1–2 cardinal signs in addition to other signs, are present.Citation38
An unusual spectrum of cancers has been observed in WS subjects who usually die at a mean age of 53–54 years from cancer or arteriosclerosis.Citation30 Controversy exists regarding the degree of brain involvement in WS.Citation1 A discussion of recent reports of cancer, atherosclerosis, and nervous system disorders in WS will be provided in the following sections.
Atherosclerosis and cancer in WS
Complications caused by atherosclerosis and cancer represent the major cause of death in WS subjects. Arteriosclerosis obliterans has been observed in more than 20% of WS subjects, and coronary heart disease in 11%–16% of them.Citation38 Less frequent are cerebral hemorrhage and cerebral infarction, observed in 2%–5% of cases.Citation38 According to recent trend analyses, atherosclerosis in WS subjects might result from either abnormal lipid metabolism, or from inflammatory mechanisms.Citation33
Concerning the cancer incidence in WS individuals, 339 (23%) out of the 1,487 WS cases described up to the end of 2008 were diagnosed with cancer,Citation33 and a nationwide epidemiological study carried out in Japan from 2009 to 2011 revealed that cancer occurred in more than 40% of the 196 analyzed WS cases.Citation38 Cancer in WS individuals often manifests with early age of onset, a high frequency of specific tumor types, including uncommon tumor types and unusual tumor sites, and with the presence of multiple tumors in individual patients.Citation39
Lauper et alCitation39 have recently provided a detailed and quantitatively rigorous view of cancer type and associated type-specific risk in WS. Frequent neoplasms included: thyroid neoplasms (16.1%), malignant melanoma (13.3%), meningioma (10.9%), soft tissue sarcomas (10.1%), hematologic/lymphoid cancers (9.3%), and osteosarcomas (7.7%). Other cancers, including nonmelanoma skin cancer, gastrointestinal cancer, ovarian cancer, genitourinary tract cancers, hepatobiliary cancer, head and neck carcinomas, and breast cancer, were observed in 3%–5% of the cases. Less frequent cancers (1%–2%) were those of the lung and central nervous system, as well as adrenocortical carcinomas.Citation39 A trend analysis in Japan revealed that the average age of onset of malignancy in WS increased from 37 years old in 1966 to 49 years old in 2008.Citation33
Nervous system disorders in WS
We recently reviewed neurological abnormalities in premature aging disorders, and they are not constant findings in WS.Citation1 Brain atrophy has been observed in 40% of WS patients, but only a few cases of dementia or peripheral neuropathy have been reported. A few cases of dementia have been recorded, but the analysis of amyloid beta pep-tide and hyperphosphorylated tau protein (the hallmarks of Alzheimer’s dementia) revealed that WS individuals do not usually appear to be susceptible to Alzheimer’s disease.Citation1 Myelopathy, demyelinization, and associated central and/or peripheral neuropathy have been described in at least six patients.Citation40–Citation44 Schizophrenia of the paranoia type was also recorded in about 10% of patients.Citation33
Genetics of WS
Typical WS is an autosomal recessive genetic disease. Therefore, the parents of a proband are obligate heterozygotes for a disease-causing mutation, and at conception each sibling of an affected individual has a 25% chance of being affected and a 50% chance of being an asymptomatic carrier. Classical WS is caused by mutations of the WRN gene on chromosome 8. The locus spans approximately 250 kb and consists of 35 exons.Citation34
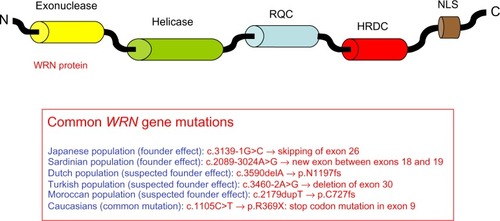
More than 70 WRN gene mutations have been found in WS patients; these are mainly nonsense mutations, insertions, and/or deletions, as well as splice mutations resulting in the production of truncated proteins lacking the nuclear localization signal, with subsequent absence of functional WRN protein in nuclei.Citation36,Citation45 The WRN protein has several functional domains (), and mutations leading to WS have been observed in all of them. There are founder mutations reported among Japanese patients (c.3139-1G>C, which results in skipping of exon 26) and in Sardinian patients (c.2089-3024A>G, which creates a new exon between exons 18 and 19).Citation30,Citation31 Potential founder mutations have been reported for Dutch (c.3590delA, p.N1197fs), Turkish (c.3460-2A>G, exon 30 deletion), and Moroccan (c.2179dupT, p.C727fs) patients.Citation36 The most common mutation in Caucasians is a stop codon mutation in exon 9 (c.1105C>T, p.R369X), accounting for approximately 20% of the mutations.Citation45 Genetic screening combined with Western blot analysis are performed as routine diagnostic tests, and shows the most frequent mutations observed by ethnic group.
Figure 2 Representation of the WRN protein and summary of the most common WRN mutations leading to Werner syndrome.
Abbreviations: RQC, RecQ C-terminal domain; HRDC, helicase and RNase D C-terminal domain; NLS, nuclear localization signal; N, amino terminal; C, carboxyl terminal.
Conclusion
Advancements in our understanding of the genetic and molecular basis of premature aging disorders have led to a better understanding of the onset and progression of the clinical manifestations. They include, for example, the recent discovery of high- and low-expressing LMNA alleles that might account for the different phenotypes observed in HGPS patients, a better characterization of the mutations leading to impaired or completely absent ZMPSTE24 protein activity, or those in the LMNA gene leading to atypical HGPS, overall accounting for a spectrum of disorders ranging from neonatal progeria to atypical WS.Citation20–Citation28
In parallel, the advancements in diagnostic examination techniques, and the creation of disease registers or databases allowing large case–control epidemiological studies in both HGPS and WS patients, have led to a better understanding of the epidemiology of disease-associated signs and symptoms. This has led, for example, to a proposed revision of the clinical diagnostic criteria for WS, to the discovery that cerebral infarctions in HGPS are more frequent than expected, and to a better understanding of cancer incidence in WS, among others.Citation12,Citation38,Citation39
Treatments for HGPS and WS are only available for the symptoms rather than for the disease itself. For example, aggressive treatment of skin ulcers, cholesterol-lowering drugs if the lipid profile is abnormal, control of type 2 diabetes mellitus, surgical treatment of cataracts, and treatment of malignancies in a standard fashion are among the available medicaments in WS, which are often accompanied by dietary/physical regimens to reduce atherosclerosis risk.Citation1
Similarly, drugs, dietary changes, and exercise are recommended for HGPS individuals to counteract atherosclerosis risk, body fat reduction, and atrophy of muscles.Citation1 Interestingly, the results of the first clinical trial in HGPS revealed that lonafarnib, a farnesyltransferase inhibitor, resulted in better hearing, improved bone structure, and led to gaining additional weight and/or to increased flexibility of the blood vessels.Citation46 In this regard, recent trend analyses in Japanese WS patients are extremely important,Citation33 as they showed a delayed onset of typical progeroid phenotypes in WS, which may be explained by the environmental changes including lifestyle and medical improvements, thereby offering a potential for interventional trials.
It is clear from that survey that, in recent years, there has been a trend toward an increase in body size in WS patients, as well as a delayed average age at onset of malignancies that follow the rapid improvement and changes in the average lifespan and lifestyle in the general population all over the world.Citation33 The future will be to combine genetic, diagnostic, and epidemiological data in order to clarify several of these gene–environment interactions and their relevance to the onset of disease-related symptoms. In addition, as recently shown by some authors studying HGPS patients,Citation28 a deeper characterization of the genetics of prematurely aged individuals might help to explain and predict the onset and progression of some of the symptoms, since several of these symptoms might result from the combined presence of different mutations in the same or different genes, thereby opening a window for personalized interventions.
Disclosure
The author reports no conflicts of interest in this work.
References
- CoppedèFPremature aging syndromeAdv Exp Med Biol201272431733122411253
- PollexRLHegeleRAHutchinson-Gilford progeria syndromeClin Genet200466537538115479179
- HutchinsonJCongenital absence of hair and mammary glands with atrophic condition of the skin and its appendages, in a boy whose mother had been almost wholly bald from alopecia areata from the age of sixMed Chir Trans188669473477
- GilfordHProgeria: a form of senilismPractitioner190473188217
- ErikssonMBrownWTGordonLBRecurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndromeNature2003423693729329812714972
- DechatTPfleghaarKSenguptaKNuclear lamins: major factors in the structural organization and function of the nucleus and chromatinGenes Dev200822783285318381888
- GoldmanRDShumakerDKErdosMRAccumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndromeProc Natl Acad Sci USA2004101248963896815184648
- HennekamRCHutchinson-Gilford progeria syndrome: review of the phenotypeAm J Med Genet A2006140232603262416838330
- GordonLBBrownWTCollinsFSHutchinson-Gilford progeria syndromePagonRAAdamMPBirdTDDolanCRFongCTStephensKGeneReviews [Internet]Seattle, WAUniversity of WashingtonSeattle19932013 [updated 2011]
- QiYCXieXHHutchinson-Gilford progeria syndrome and its relevance to cardiovascular diseases and normal agingBiomed Environ Sci201326538238923611131
- Gerhard-HermanMSmootLBWakeNMechanisms of premature vascular aging in children with Hutchinson-Gilford progeria syndromeHypertension2012591929722083160
- SilveraVMGordonLBOrbachDBCampbellSEMachanJTUllrichNJImaging characteristics of cerebrovascular arteriopathy and stroke in Hutchinson-Gilford progeria syndromeAJNR Am J Neuroradiol20133451091109723179651
- SchmidtENilssonOKoskelaAExpression of the Hutchinson-Gilford progeria mutation during osteoblast development results in loss of osteocytes, irregular mineralization, and poor biomechanical propertiesJ Biol Chem201228740335123352222893709
- ClevelandRHGordonLBKleinmanMEA prospective study of radiographic manifestations in Hutchinson-Gilford progeria syndromePediatr Radiol20124291089109822752073
- UllrichNJSilveraVMCampbellSEGordonLBCraniofacial abnormalities in Hutchinson-Gilford progeria syndromeAJNR Am J Neuroradiol20123381512151822460337
- GuardianiEZalewskiCBrewerCOtologic and audiologic manifestations of Hutchinson-Gilford progeria syndromeLaryngoscope2011121102250225521898437
- KalilKAFargalleyHSHypoparathyroidism in an Egyptian child with Hutchinson-Gilford progeria syndrome: a case reportJ Med Case Rep201261722251708
- CoppedèFMiglioreLDNA repair in premature aging disorders and neurodegenerationCurr Aging Sci20103131920298165
- De Sandre-GiovannoliABernardRCauPLamin a truncation in Hutchinson-Gilford progeriaScience20033005628205512702809
- MoulsonCLFongLGGardnerJMIncreased progerin expression associated with unusual LMNA mutations causes severe progeroid syndromesHum Mutat200728988288917469202
- FukuchiKKatsuyaTSugimotoKLMNA mutation in a 45 year old Japanese subject with Hutchinson-Gilford progeria syndromeJ Med Genet2004415e6715121795
- ShalevSADe Sandre-GiovannoliAShaniAALevyNAn association of Hutchinson-Gilford progeria and malignancyAm J Med Genet A2007143A161821182617618517
- DoubajYDe Sandre-GiovannoliAVeraEVAn inherited LMNA gene mutation in atypical Progeria syndromeAm J Med Genet A2012158A112881288722991222
- BarrowmanJWileyPAHudon-MillerSEHrycynaCAMichaelisSHuman ZMPSTE24 disease mutations: residual proteolytic activity correlates with disease severityHum Mol Genet201221184084409322718200
- ChenLLeeLKudlowBALMNA mutations in atypical Werner’s syndromeLancet2003362938244044512927431
- KaneMSLindsayMEJudgeDPLMNA-associated cardiocutaneous progeria: An inherited autosomal dominant premature aging syndrome with late onsetAm J Med Genet A201316171599161123666920
- ConneelyKNCapellBCErdosMRHuman longevity and common variations in the LMNA gene: a meta-analysisAging Cell201211347548122340368
- RodríguezSErikssonMLow and high expressing alleles of the LMNA gene: implications for laminopathy disease developmentPLoS ONE201169e2547221980471
- JacobKNGargALaminopathies: multisystem dystrophy syndromesMol Genet Metab200687428930216364671
- GotoMClinical aspects of Werner’s syndrome: its natural history and the genetics of the diseaseLebelMMolecular Mechanisms of Werner’s SyndromeNew York, NYKluver Academic Plenum Publishers2004111
- MasalaMVScapaticciSOlivieriCEpidemiology and clinical aspects of Werner’s syndrome in North Sardinia: description of a clusterEur J Dermatol200717321321617478382
- WernerOOn Cataract in Conjunction with Scleroderma [dissertation]Kiel, GermanySchmidt and Klaunig1904
- GotoMIshikawaYSugimotoMFuruichiYWerner syndrome: a changing pattern of clinical manifestations in Japan (1917∼2008)Biosci Trends201371132223524889
- YuCEOshimaJFuYHPositional cloning of the Werner’s syndrome geneScience199627252592582628602509
- MuftuogluMOshimaJvon KobbeCChengWHLeistritzDFBohrVAThe clinical characteristics of Werner syndrome: molecular and biochemical diagnosisHum Genet2008124436937718810497
- FriedrichKLeeLLeistritzDFWRN mutations in Werner syndrome patients: genomic rearrangements, unusual intronic mutations and ethnic-specific alterationsHum Genet2010128110311120443122
- MelcherRvon GolitschekRSteinleinCSpectral karyotyping of Werner syndrome fibroblast culturesCytogenet Cell Genet2000911–418018511173853
- TakemotoMMoriSKuzuyaMDiagnostic criteria for Werner syndrome based on Japanese nationwide epidemiological surveyGeriatr Gerontol Int201313247548122817610
- LauperJMKrauseAVaughanTLMonnatRJSpectrum and risk of neoplasia in Werner syndrome: a systematic reviewPLoS ONE201384e5970923573208
- AndersonNEHaasLFNeurological complications of Werner’s syndromeJ Neurol2003250101174117814586597
- MalandriniADottiMTVillanovaMBattistiCFedericoANeurological involvement in Werner’s syndrome: clinical and biopsy study of a familial caseEur Neurol200044318718911053972
- UmeharaFAbeMNakagawaMWerner’s syndrome associated with spastic paraparesis and peripheral neuropathyNeurology1993436125212548170578
- JustACanapleSJolyHPiussanCRosaAComplications neurologiques dans un cas de syndrome de Werner [Neurologic complications in a case of Werner syndrome.]Rev Neurol (Paris)199615210634636 French9033957
- KawamuraHMoriSMuranoSYokoteKTamuraKSaitoY[Werner’s syndrome associated with progressive subcortical vascular encephalopathy of the Binswanger type.]Nihon Ronen Igakkai Zasshi1999369648651 Japanese10572450
- HisamaFMKubischCMartinGMOshimaJClinical utility gene card for: Werner syndromeEur J Hum Genet2012205
- GordonLBKleinmanMEMillerDTClinical trial of a farnesyltransferase inhibitor in children with Hutchinson-Gilford progeria syndromeProc Natl Acad Sci USA201210941166661667123012407