Abstract
Background and aim
Amyloid-beta (Aβ) peptide is reported to initiate flexible inflammation in the hippocampus of the human brain in Alzheimer’s disease (AD). The present study aimed to investigate the possible effects of atorvastatin on the production of inflammation cytokines in the hippocampus and the potential impacts on behavioral ability, in an AD model.
Methods
We firstly established AD rat models using intracerebroventricular injection of Aβ1-42. A Morris water maze was also performed to determine the spatial learning and memory ability in the AD models. Intracellular staining of interleukin (IL)-1β, IL-6, and tumor necrosis factor alpha was determined using immunohistochemical staining at 6 hours and day 7 after the injection of Aβ.
Results
The escape latency of the atorvastatin-treated AD group (5 mg/kg/d) was significantly shorter than that of AD group on day 3 (41 ± 1.05 seconds versus 47 ± 1.05 seconds, P < 0.01) and day 4 (34 ± 1.25 seconds versus 43 ± 1.01 seconds, P < 0.01) after the beginning of the training. Furthermore, the atorvastatin-treated AD group displayed a significant higher mean number of annulus crossings than did the AD group (2.9 ± 0.5 versus 2.4 ± 0.9, P < 0.05). Fewer injured nerve cells and proliferated glial cells were also demonstrated in the atorvastatin-treated AD group than in the AD group. Of great importance, we demonstrated that IL-1β, IL-6, and tumor necrosis factor alpha were significantly decreased in the atorvastatin-treated AD group than that in the AD group.
Conclusion
Atorvastatin might attenuate the damage of nerve cells and improve learning and memory ability by inhibiting inflammatory response in the progression of AD.
Introduction
Alzheimer’s disease (AD) is a progressive neurodegenerative condition which is characterized by the extraneuronal deposits of beta-amyloid (Aβ) fibrils and intraneuronal accumulations of hyperphosphorylated tau protein.Citation1 In the progression of AD, it has been demonstrated that accumulation and aggregation of Aβ peptide in the hippocampus of the brain usually results in the activation of glial cells which, in turn, initiates a neuroinflammatory response, involving reactive oxygen intermediates and inflammatory cytokines, including interleukin (IL)-1β, IL-6, and tumor necrosis factor (TNF-α).Citation1,Citation2 In addition, soluble Aβ is also reported to induce inflammation.Citation3
Experimental and clinical data demonstrate that cholesterol is a possible factor in the etiology of ADCitation4 Cholesterol-lowering agents, particularly 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitors (also known as statins), have been reported to be capable of increasing the ratio of alpha to beta secretase activity and then increasing the concentrations of extracellular Aβ.Citation5–Citation7 Furthermore, accumulating clinical epidemiological data from various populations has showed that statin users have had low incidence of AD.Citation8–Citation11 This low incidence of AD in statins users was previously thought to be attributed to the cholesterol-lowering activity of statins. Currently the non-cholesterol-lowering actions of statins, including their action on nitric oxide, platelet adhesion, and antiinflammatory action, have been reported.Citation12 In addition, statins can also increase endothelial nitric oxide synthase (eNOS), and reduce endothelin-1, thereby resulting in relaxation of vascular smooth muscles and leading to vasodilatation.Citation13,Citation14 Interestingly, inflammatory response has been demonstrated to be involved in the activation of plaque-associated microglia in the progression of AD. The activated microglias are also capable of releasing a wide range of proinflammatory factors and then exacerbating neuronal death.Citation15,Citation16 Therefore, it is speculated that the antiinflammatory activity of statins might partly contribute to the low incidence of AD in statins users.
Simvastatin has been demonstrated to reduce levels of Aβ42 and Aβ40 in vitro and in vivo.Citation17 Atorvastatin is a member of the statin family, which is mainly prescribed for the treatment of hypercholesterolemia and has been shown to have potential beneficial therapeutic anti-inflammatory effects.Citation18 Therefore, we speculated that atorvastatin might attenuate learning and memory ability in an AD model, through the inhibition of inflammatory response. In this present study, we then mainly intended to investigate the possible effects of atorvastatin on hippocampal inflammation and behavioral ability, including learning and memory, in an AD rat model induced by Aβ1-42.
Materials and methods
Animal treatment
A total of 60 adult male Wistar rats (250–350 g), purchased from the Experimental Animal Center of Shandong University (Shandong, People’s Republic of China), were included in this study. The rats were housed in groups of five, had free access to food and water, and were maintained under veterinary supervision at an ambient temperature of 22°C–23°C, under a 12:12 hour light/dark cycle. All the rats received human care, and the experiment was approved by the Animal Care and Use Committee of Shandong University, and every effort was made to minimize stress to the animals. The rats were adapted for 2 weeks to the above conditions prior to the present experiment.
All the rats were randomly classified into four groups: the control group (Group 1); the AD group (Group 2), the atorvastatin control group (Group 3); and the atorvastatintreated AD group (Group 4). A Morris water maze (MWM) was performed at the beginning of the study and there was no significant difference among the four groups. Groups 3 and 4 were treated with atorvastatin for 3 weeks, and the other two groups were treated with saline. After 3 weeks, Groups 2 and 4 were injected intracerebroventricularly with Aβ1-42, and the other two groups with saline. Brain tissue was obtained for pathological observation and intracellular staining at 6 hours (n = 5) and on day 7 after Aβ1-42 injection (n = 5). An MWM (n = 5) was performed to identify learning and memory ability, on day 7 after the establishment of the AD model.
Materials
Aβ1-42 (Sigma-Aldrich, St Louis, MO, USA) was dissolved in sterile saline at a concentration of 2 g/L and stored at –20°C. To obtain the neurotoxic form of Aβ1-42, the peptide solution was placed in an incubator at 37°C for 7 days. Rabbit anti-IL-1β, goat anti-IL-6, and goat anti-TNF-α polyclonal antibodies were obtained from Santa Cruz Biotechnology Inc (Santa Cruz, CA, USA). Biotin conjugated anti-rabbit or anti-goat secondary antibody was purchased from Beijing Zhongshan Biotechnology Co., Ltd (Beijing, People’s Republic of China).
Induction of cognitive dysfunction by injection of Aβ1-42 and treatment with atorvastatin
The AD model was induced with the injection of Aβ1-42 into the ventricle (2.5 mm posterior and 0.5 mm lateral to the bregma). A microsyringe with a stainless steel needle was used for this experiment. Rats were anesthetized with sodium pentobarbital (40 mg/kg) and placed in a stereotaxic apparatus. Then, 5 mL of peptide (10 μg) or vehicle control was delivered gradually within 3 minutes, and the syringe was kept at the site for 2 minutes following the injection.
Atorvastatin, 5 mg/kg per day (Lipitor®; Pfizer Inc, New York, NY, USA) and saline (2 mL) were mixed with regular food via intragastric administration, and rats were examined daily and weighed at intervals throughout the study. The dose of atorvastatin was decided on the basis of previous findings;Citation19,Citation20 this was higher than the human dose (1.1 mg/kg per day) recommended for the treatment of hypercholesterolemiaCitation21 but was consistent with the pharmacokinetic data reporting a higher metabolic rate of the drug in rodents.Citation22
Morris water maze
All the rats were trained on a MWM test on day 7 after the injection of Aβ1-42. The apparatus consisted of a large circular pool, approximately 1.0 meter in diameter, containing water, at around 25°C, that was made opaque by the addition of milk to help hide the submerged platform. The movements of the animal in the tank were monitored with a video tracking system (EthoVision, Noldus Information Technology, Wageningen, The Netherlands).
Reference memory test
For each training trial, the rat was put into the pool at one of the five positions, the sequence of the positions being selected randomly. The platform was located a constant position throughout the test period in the middle of one quadrant and equidistant from the center and edge of the pool. In each training session, the latency to escape onto the hidden platform was recorded. If the rat found the platform, it was allowed to remain there for 10 seconds and was then returned to its home cage. If the rat was unable to find the platform within 60 seconds, the training was terminated and a maximum score of 60 seconds was assigned. Training was conducted for four consecutive days, four times per day, from day 7 to 10, after injection of Aβ1-42.
Probe test
On day 11 after injection of Aβ1-42, a single probe trial was conducted. The platform was removed from the pool, and each rat was allowed to swim for 60 seconds in the maze. The number of times the rat crossed the annulus where the platform had been located was recorded.
Working memory (repeated acquisition) test
A working memory test was conducted on three consecutive days from day 11 to 13 and consisted of five trials per day. The working memory test was procedurally similar to reference memory test except that the platform location was changed daily. The first trial of the day was an informative sample trial in which the rat was allowed to swim to the platform in its new location. Spatial working memory was regarded as the mean escape latency of the second to fifth trials.Citation23
Pathological features
Cardiovascular perfusion fixation was carried out after behavioral test. The rats, anesthetized by celiac injection of 10% chloral hydrate (3 mL/kg), were perfused with 100 mL chilled 0.9% saline first and then fixed with 150 mL paraformaldehyde (4%, 4°C). The brains were taken out quickly on an icy table and then put into a paraformaldehyde solution (4%) for fixation. Sequential tissue sections (5 μm thick) were made along the coronal profile, after fixation and paraffin embedment. Hippocampal sections were put on clean slides treated with poly-D-lysine and baked for 2 hours at 60°C, then kept in a refrigerator at 4°C, for hematoxylin and eosin (HE) staining. Structural features were observed at 6 hours and on day 7, after Aβ1-42 injection.
Immunohistochemistry procedure
IL-1β, IL-6, and TNF-α proteins expressed in the brain tissue were detected by immunostaining, according to the standard streptavidin-biotin-peroxidase technique. Briefly, the paraffin wax-embedded sections were dewaxed and incubated with 3% H2O2 for 5 minutes, to inactivate the native peroxidase. After rinsing with distilled water, the sections were kept in phosphate-buffered saline (PBS) for 5 minutes and incubated with primary antibody diluent for 10 minutes. After incubation with primary antibody diluent, the sections were rinsed three times for five minutes with PBS and then incubated with secondary antibody diluent for 30 minutes. The sections were rinsed three times for 3 minutes with PBS before incubation with streptavidin-HRP complex and reaction was with 3,3′-diaminobenzidine (DAB) solution for 10 minutes after being rinsed another three times for 5 minutes with PBS. The sections were enveloped with gelatin and observed under the microscope. For semiquantitative immunohistochemistry, the number of positive cells was counted by an investigator blinded to the experimental groups. Under the microscope, the counting area for hippocampus CA1 area of each section was exactly the size of a complete field at ×200 magnification. Only the positive cells inside the pyramidal layer were counted. An average of the serial four slices was taken from each individual rat. Four rats were taken from each group.
Statistical analysis
Data was expressed as mean ± standard derivation (SD) for each individual. Statistical analysis of the data for multiple comparisons was performed by one way analysis of variance (ANOVA) with a post hoc test. A level of P < 0.05 was accepted as statistically significant.
Results
Effects of atorvastatin on learning and memory
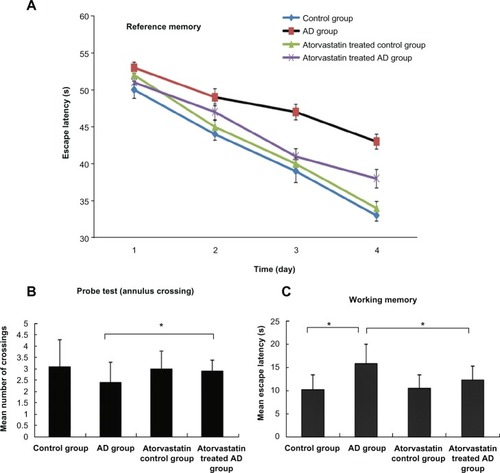
The spatial learning and memory ability of rat models were determined using the following parameters: escape latency onto a hidden platform, mean number of annulus crossings, and mean escape latency. We obviously found that the escape latencies of the AD rat model after 2–4 days of training were significantly longer than the two groups without AD. demonstrates that the escape latency of the atorvastatin-treated AD group was significantly shorter than AD group on day 3 (41 ± 1.05 seconds versus 47 ± 1.05 seconds, P < 0.01) and day 4 (34 ± 1.25 seconds versus the beginning of the training 43 ± 1.01 seconds, P < 0.01). In the 60 second probe test, the mean number of annulus crossings of the atorvastatin-treated AD group showed significant higher times of annulus crossing than did the AD group (2.9 ± 0.5 versus 2.4 ± 0.9, P < 0.05) (). In , working memory was assessed as the mean escape latency of the second to the fifth trial for 3 days. Of great interest, the atorvastatin-treated AD group showed a significant decrease in mean escape latency from the AD group (12.3 ± 3 seconds versus 15.8 ± 4.2 seconds, P < 0.05). These results strongly suggested that atorvastatin might be capable of attenuating the impaired spatial learning and memory in AD rat models.
Figure 1 Atorvastatin improved the spatial learning and memory of rats impaired by Aβ1-42, using the Morris water maze test. (A) Escape latency onto a hidden platform; (B) probe test; and (C) working memory.
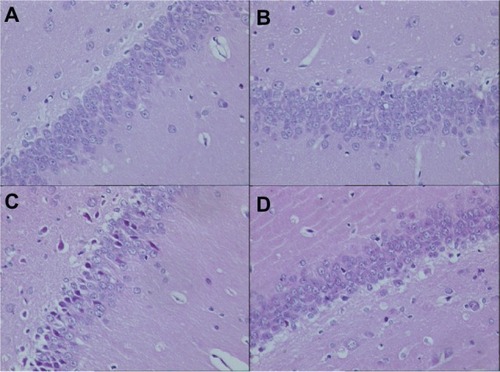
Effects of atorvastatin on pathological changes in rats’ hippocampus
In this present study, HE staining was used for identifying the different macroscopic pathological features in the hippocampus of AD rat models. On day 7 after Aβ1-42 injection, the cellular nuclei were clear and homogeneously stained in the groups without atorvastatin treatment ( and C). Furthermore, sparsely formed cells and deeply stained nuclei were obviously demonstrated in the AD group (). However, in the atorvastatin-treated AD group (), the neurons and the nuclei just looked the same as in the control group without Aβ1-42 injection.
Figure 2 Pathological changes in the hippocampus of rats in each group on day 7 after Aβ injection (original magnification × 400). (A) Control group; (B) atorvastatin control group; (C) AD group; and (D) atorvastatin-treated AD group.
Effects of atorvastatin on the expression of proinflammatory factors
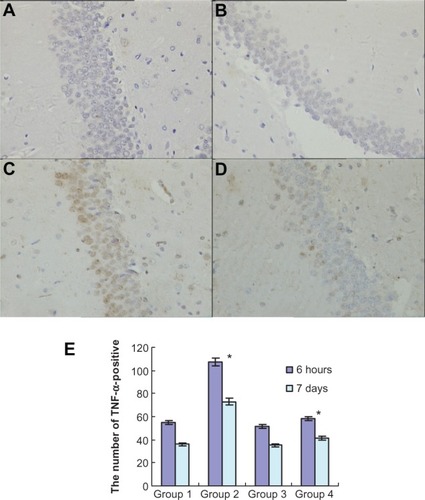
To investigate the effects of atorvastatin on hippocampal inflammation, we determined the intracellular staining of certain proinflammatory factors, including IL-1β, IL-6, and TNF-α, using an immunohistochemical method, at 6 hours and 7 days after the injection of Aβ. Generally, intracellular staining of IL-1β, IL-6, and TNF-α was mostly located in the cell plasma. The results showed that there was no obvious staining of IL-1β, IL-6, or TNF-α in the hippocampus of the control group (, , and ). However, intracellular staining of IL-1β, IL-6, and TNF-α were increased significantly (, , and ) in the Aβ-treated group over that in the control and atorvastatin control groups. There were significant differences in the intracellular staining of IL-1β, IL-6, and TNF-α in the atorvastatin control group and the control group (, , and , , ). Of great importance, we demonstrated that IL-1β, IL-6, and TNF-α were significantly decreased in the atorvastatin-treated AD group than that in the AD group (, , , and , , ).
Figure 3 Atorvastatin attenuated IL-1β expression in the hippocampus of Aβ1-42-treatedrats. The upper panel shows IL-1 β-positive cells in the rat hippocampus, detected by immunohistochemistry on day 7 after Aβ injection (original magnification x 400). (A) Control group (Group 1); (B) atorvastatin control group (Group 2); (C) AD group (Group 3); and (D) atorvastatin-treated AD group (Group 4). (E) The lower panel shows the counted number of IL-1 β-positive cells in rat hippocampus.
Notes: Data are the mean ± SD of each individuals. *P < 0.01.
Abbreviations: Aβ, amyloid-beta (peptide); IL, interleukin; AD, Alzheimer’s disease.
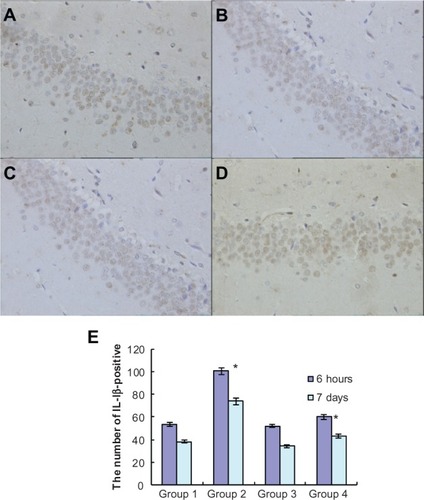
Figure 4 Atorvastatin attenuated IL-6 expression in the hippocampus of Aβ1-42-treated rats. The upper panel shows IL-6-positive cells in the rat hippocampus, detected by immunohistochemistry on day 7 after Aβ injection (original magnification x 400). (A) Control group (Group 1); (B) atorvastatin control group (Group 2); (C) AD group (Group 3); and (D) atorvastatin-treated AD group (Group 4). (E) The lower panel shows the counted number of IL-6-positive cells in rat hippocampus.
Notes: Data are the mean ± SD of each individuals. *P < 0.01.
Abbreviations: IL, interleukin; Aβ, amyloid-beta (peptide); AD, Alzheimer’s disease.
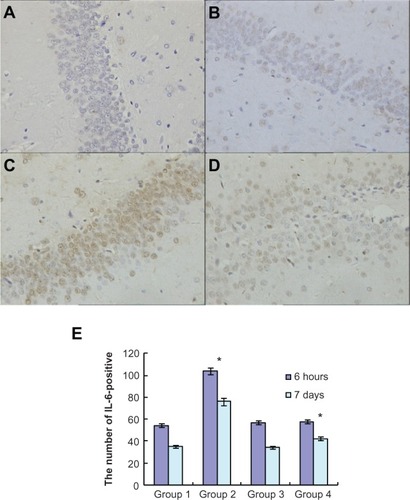
Figure 5 Atorvastatin attenuated TNF-α expression in the hippocampus of Aβ1-42-treated rats. The upper panel shows TNF-α-positive cells in the rat hippocampus, detected by immunohistochemistry on day 7 after Aβ injection (original magnification × 400). (A) Control group (Group 1); (B) atorvastatin control group (Group 2); (C) AD group (Group 3); and (D) atorvastatin-treated AD group (Group 4). (E) The lower panel shows the counted number of TNF-α-positive cells in rat hippocampus.
Abbreviations: TNF-α, tumor necrosis factor alpha; Aβ, amyloid-beta (peptide); AD, Alzheimer’s disease.
Discussion
The clinical data demonstrated that statins might reduce the risk rate for the incidence of AD. Meanwhile, the cholesterol-lowering activity and anti-inflammatory ability of the statin family have been considered to provide rationale for this hypothesis.Citation10,Citation19 In this present study, we firstly established the AD rat model using Aβ1-42 injection and then aimed to investigate the possible effects of atorvastatin on the inflammation and on behavioral ability, including learning and memory. Our results demonstrated that the escape latency of the atorvastatin-treated AD group was significantly shorter than in the AD group on the third and fourth day after the beginning of the training. Furthermore, the atorvastatintreated AD group displayed a significant higher mean number of annulus crossings than did the AD group. Fewer injured nerve cells and proliferated glial cells were also found in the atorvastatin-treated AD group than in the AD group. Of great importance, we demonstrated that IL-1β, IL-6, and TNF-α were significantly decreased in the atorvastatin-treated AD group than that in the AD group. Our results strongly suggested that atorvastatin might attenuate the damage of nerve cells and improve learning and memory ability by inhibiting inflammatory response in the progression of AD.
In our present study, we found that the intracellular expressions of IL-1β, IL-6, and TNF-α was significantly increased after the injection of Aβ. Previous studies also demonstrated that increased inflammatory components contributed to the progression of AD.Citation24 It is therefore hypothesized that inflammatory components could be involved in the activation of plaque-associated microglia. It was also demonstrated that inflammation might act to augment disease progression via its ability to stimulate neuronal Aβ production in the progression of AD, and to facilitate amyloid deposition.Citation25–Citation28 However, the exact mechanism underlying the inflammatory response action on the progression of AD is still obscure and needs to be further studied.
The hypothesis that statin treatment may be one independent factor in the incidence of AD has been strongly suggested. Wolozin et alCitation29 performed a cross-sectional design and reported that statin therapy provided a 60%–73% benefit in the risk of AD, compared with that in the general population. Subsequent retrospective studies and prospective clinical investigations also provided evidence to support this hypothesis.Citation30,Citation31 The epidemiological findings have demonstrated that high-cholesterol diets result in the exacerbation of Aβ deposition, and this effect could be reversed by statin treatment.Citation32,Citation33 However, the exact molecular mechanism underlying the statin association with low incidence of AD has not been well demonstrated.
Statin treatment has been shown to reduce the levels of matrixmetalloproteinase-9, TNF-α, and monocyte chemotactic protein-1 and to decrease the activities of NF-κB, the NADPH oxidase complex in both vascular and myeloid-lineage cells.Citation34,Citation35 Statins have also been shown to protect neurons from excitotoxic injury.Citation36 The finding of potent anti-inflammatory effects of statins provides a new dimension in the action of statins in the AD brain. There is a strong rationale for why anti-inflammatory therapies may be efficacious in the treatment of AD. In the present study, our results showed that atorvastatin could attenuate the Aβ-stimulated injury to learning and memory, and partly inhibit the inflammatory responses in the hippocampus of the rat brain. These results strongly support our hypothesis that atorvastatin could exert non-cholesterol-lowering activity in AD progression. Notably, our results must be interpreted with great caution. The effects of atorvastatin treatment seemed dramatic and were surprising. Perhaps the dramatic differences come from the small number of rats, selection bias, the strains of the rats, and so on. However, the anti-inflammatory role of atorvastatin provided a potential therapy for the prevention of AD and this issue must be studied in future research.
There are some limitations in this present study. First, we only observed the positive staining of inflammatory cytokines in the hippocampus of an AD model. Other cells that stain positive for IL-1β, IL-6, and TNF-α must be identified using markers such as Iba-1 for microglia or glial fibrillary acidic protein (GFAP) for astrocytes. Second, we performed immunohistological staining to determine the presence of an inflammatory response in the brain. In future work, better quantitative assays are necessary such as enzyme-linked immunosorbent assay (ELISA), Western blot, or reverse transcription polymerase chain reaction (RT-PCR). However, our results still provide strong evidence that atorvastatin might attenuate the damage of nerve cells and improve learning and memory ability by inhibiting inflammatory response in the progression of AD.
Conclusion
Our present study aimed to investigate the possible effects of atorvastatin on inflammation and on behavioral ability, including learning and memory. Our results firstly demonstrated improved learning and memory ability under the treatment of atorvastatin, using AD rat models. We then found that IL-1β, IL-6, and TNF-α were significantly decreased in the atorvastatin-treated AD group than that in the AD group. Our results strongly suggested that atorvastatin might attenuate the damage of nerve cells and improve learning and memory ability, by inhibiting inflammatory response in the progression of AD. The potential therapeutic role of atorvastatin in the prevention of AD should be studied in future research.
Acknowledgment
This work was supported by grants from National Natural Science Foundation Shandong Province (Y2008C103) and Jinan Planning Project of Science and Technology (200807032).
Disclosure
The authors declare that they have no competing financial interests.
References
- MuckeLNeuroscience: Alzheimer’s diseaseNature2009461726689589719829367
- VetrivelKSThinakaranGMembrane rafts in Alzheimer’s disease beta-amyloid productionBiochim Biophys Acta20101801886086720303415
- WhiteJAManelliAMHolmbergKHVan EldikLJLaduMJDifferential effects of oligomeric and fibrillar amyloid-beta 1–42 on astrocyte-mediated inflammationNeurobiol Dis200518345946515755672
- MartinsIJBergerTSharmanMJVerdileGFullerSJMartinsRNCholesterol metabolism and transport in the pathogenesis of Alzheimer’s diseaseJ Neurochem200911161275130820050287
- BuxbaumJDGeoghagenNSFriedhoffLTCholesterol depletion with physiological concentrations of a statin decreases the formation of the Alzheimer amyloid Abeta peptideJ Alzheimers Dis20013222122912214063
- ParsonsRBPriceGCFarrantJKSubramaniamDAdeagbo-SheikhJAustenBMStatins inhibit the dimerization of beta-secretase via both isoprenoid- and cholesterol-mediated mechanismsBiochem J2006399220521416803455
- ColeSLGrudzienAManhartIOKellyBLOakleyHVassarRStatins cause intracellular accumulation of amyloid precursor protein, beta-secretase-cleaved fragments, and amyloid beta-peptide via an isoprenoid-dependent mechanismJ Biol Chem200528019187551877015718241
- FonsecaACResendeROliveiraCRPereiraCMCholesterol and statins in Alzheimer’s disease: current controversiesExp Neurol2010223228229319782682
- IsingriniEDesmidtTBelzungCCamusVEndothelial dysfunction: A potential therapeutic target for geriatric depression and brain amyloid deposition in Alzheimer’s disease?Curr Opin Investig Drugs20091014655
- EvansBAEvansJEBakerS PLong-term statin therapy and CSF cholesterol levels: implications for Alzheimer’s diseaseDement Geriatr Cogn Disord200927651952419478483
- ReidPCUranoYKodamaTHamakuboTAlzheimer’s disease: cholesterol, membrane rafts, isoprenoids and statinsJ Cell Mol Med200711338339217635634
- Charlton-MenysVDurringtonPNSqualene synthase inhibitors: clinical pharmacology and cholesterol-lowering potentialDrugs2007671111617209661
- HessDCDemchukAMBrassLMYatsuFMHMG-CoA reductase inhibitors (statins): a promising approach to stroke preventionNeurology200054479079610690964
- KaesemeyerWHCaldwellRBHuangJCaldwellRWPravastatin sodium activates endothelial nitric oxide synthase independent of its cholesterol-lowering actionsJ Am Coll Cardiol19993312342419935036
- CombsCKJohnsonDEKarloJCCannadySBLandrethGEInflammatory mechanisms in Alzheimer’s disease: inhibition of beta-amyloid-stimulated proinflammatory responses and neurotoxicity by PPARgamma agonistsJ Neurosci200020255856710632585
- YatesSLBurgessLHKocsis-AngleJAmyloid beta and amylin fibrils induce increases in proinflammatory cytokine and chemokine production by THP-1 cells and murine microgliaJ Neurochem20007431017102510693932
- BoimelMGrigoriadisNLourbopoulosAStatins reduce the neurofibrillary tangle burden in a mouse model of tauopathyJ Neuropathol Exp Neurol200968331432519225406
- FassbenderKSimonsMBergmannCSimvastatin strongly reduces levels of Alzheimer’s disease beta-amyloid peptides Abeta 42 and Abeta 40 in vitro and in vivoProc Natl Acad Sci U S A200198105856586111296263
- PrinceSEDaselaarSMCabezaRNeural correlates of relational memory: successful encoding and retrieval of semantic and perceptual associationsJ Neurosci20052551203121015689557
- YoussefSStüveOPatarroyoJCThe HMG-CoA reductase inhibitor, atorvastatin, promotes a Th2 bias and reverses paralysis in central nervous system autoimmune diseaseNature20024206911788412422218
- BerniniFPoliAPaolettiRSafety of HMG-CoA reductase inhibitors: focus on atorvastatinCardiovasc Drugs Ther200115321121811713888
- DostalLAWhitfieldLRAndersonJAFertility and general reproduction studies in rats with the HMG-CoA reductase inhibitor, atorvastatinFundam Appl Toxicol19963222852928921332
- JhooJHKimHCNabeshimaTBeta-amyloid (1–42)-induced learning and memory deficits in mice: involvement of oxidative burdens in the hippocampus and cerebral cortexBehav Brain Res2004155218519615364477
- SwardfagerWLanctôtKRothenburgLWongACappellJHerrmannNA meta-analysis of cytokines in Alzheimer’s diseaseBiol Psychiatry2010681093094120692646
- BlaskoIApochalABoeckGHartmannTGrubeck-LoebensteinBRansmayrGIbuprofen decreases cytokine-induced amyloid beta production in neuronal cellsNeurobiol Dis2001861094110111741404
- SastreMDewachterILandrethGENonsteroidal anti-inflammatory drugs and peroxisome proliferator-activated receptor-gamma agonists modulate immunostimulated processing of amyloid precursor protein through regulation of beta-secretaseJ Neurosci200323309796980414586007
- QiaoXCumminsDJPaulSMNeuroinflammation-induced acceleration of amyloid deposition in the APPV717F transgenic mouseEur J Neurosci200114347448211553297
- GuoJTYuJGrassDde BeerFCKindyMSInfammation-dependent cerebral deposition of serum amyloid a protein in a mouse model of amyloidosisJ Neurosci200222145900590912122052
- WolozinBKellmanWRuosseauPCelesiaGGSiegelGDecreased prevalence of Alzheimer disease associated with 3-hydroxy-3-methyglutaryl coenzyme A reductase inhibitorsArch Neurol200057101439144311030795
- RockwoodKKirklandSHoganDBUse of lipid-lowering agents, indication bias, and the risk of dementia in community-dwelling elderly peopleArch Neurol200259222322711843693
- CaballeroJNahataMDo statins slow down Alzheimer’s disease? A reviewJ Clin Pharm Ther200429320921315153082
- RefoloLMPappollaMALaFrancoisJA cholesterol-lowering drug reduces beta-amyloid pathology in a transgenic mouse model of Alzheimer’s diseaseNeurobiol Dis20018589089911592856
- PetanceskaSSDeRosaSOlmVStatin therapy for Alzheimer’s disease: will it work?J Mol Neurosci2002191–215516112212773
- GripOJanciauskieneSLindgrenSAtorvastatin activates PPAR-gamma and attenuates the inflammatory response in human monocytesInfamm Res20025125862
- WongBLummaWCSmithAMSiskoJTWrightSDCaiTQStatins suppress THP-1 cell migration and secretion of matrix metalloproteinase 9 by inhibiting geranylgeranylationJ Leukoc Biol200169695996211404382
- ZaccoATogoJSpenceK3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors protect cortical neurons from excitotoxicityJ Neurosci20032335111041111114657168