
Abstract
Soft-tissue sarcomas are rare malignant tumors arising from connective tissues and have an overall incidence of about five per 100,000 per year. While this diverse family of malignancies comprises over 100 histological subtypes and many molecular aberrations are prevalent within specific sarcomas, very few are therapeutically targeted. Instead of utilizing molecular signatures, first-line sarcoma treatment options are still limited to traditional surgery and chemotherapy, and many of the latter remain largely ineffective and are plagued by disease resistance. Currently, the mechanism of sarcoma oncogenesis remains largely unknown, thus necessitating a better understanding of pathogenesis. Although substantial progress has not occurred with molecularly targeted therapies over the past 30 years, increased knowledge about sarcoma biology could lead to new and more effective treatment strategies to move the field forward. Here, we discuss biological advances in the core molecular determinants in some of the most common soft-tissue sarcomas – liposarcoma, angiosarcoma, leiomyosarcoma, rhabdomyosarcoma, Ewing’s sarcoma, and synovial sarcoma – with an emphasis on emerging genomic and molecular pathway targets and immunotherapeutic treatment strategies to combat this confounding disease.
Introduction
Soft-tissue sarcoma (STS) is a diverse group of rare cancers that arise from pathological transformations in the mesenchyme, which is the mesodermal portion of the embryo that develops into connective and skeletal tissues. These rare cancers account for <1% of all adult malignancies, and an estimated 12,000 new cases of STS are diagnosed in the US each year, with approximately 5,000 deaths.Citation1,Citation2 While the exact cause of carcinogenesis has remained elusive and these cancers can arise from any body part, most STSs are diagnosed in the extremities (59.5%), followed by the trunk (17.9%).Citation3 At the time of initial diagnosis, distant metastases are rarely present, but blood is the most common route for the disease to spread, most frequently to the lungs.Citation4
Current treatments for STS often involve multiple modalities, including surgery, radiation, and chemotherapy. There are a few US Food and Drug Administration (FDA)-approved chemotherapeutic drugs for treating STS, such as eribulin, trabectedin, ifosfamide, anthracyclines, and taxanes, but toxicity and partial responses remain significant limitations. Little progress has been made with respect to targeted therapeutics. The 5-year overall survival rate for STS is 90% for stage I, 81% for stage II, and 56% for stage III.Citation5 Beyond TNM stage and histologic grade,Citation5,Citation6 additional prognostic factors include surgical margins, age, anatomic site, and histologic subtype.Citation7
According to the World Health Organization classification, over 100 distinct histological subtypes have been categorized.Citation8 Given this diversity, the appropriate course of treatment could be guided by a better understanding of the disease pathobiology at the molecular level. In this review, we focus on several of the most common histologic subtypes in adults: liposarcoma (LPS), angiosarcoma, leiomyosarcoma (LMS), rhabdomyosarcoma (RMS), Ewing’s sarcoma (ES), and synovial sarcoma (SS). We summarize current knowledge and advances in STS biology in terms of molecular pathways and genomics ( and ). We also consider translational implications of new targeted and immunotherapeutic strategies under investigative development that could potentially permit longer survival and a better quality of life for those with STS ().
Table 1 Soft-tissue sarcoma genomic landscape
Table 2 Soft-tissue sarcoma targets, therapeutics, and clinical status
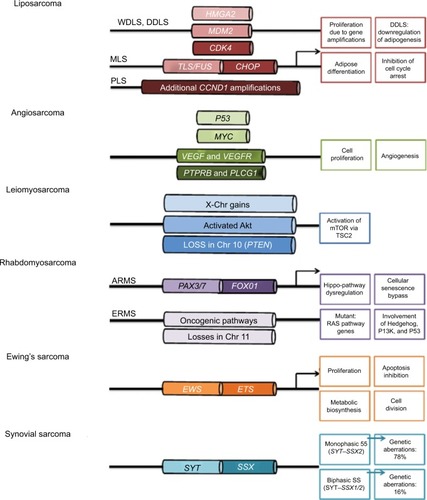
Figure 1 Genomic changes in soft-tissue sarcoma.
Notes: Liposarcomas consist of four subtypes: well-differentiated liposarcoma (WDLS), dedifferentiated liposarcoma, myxoid liposarcoma (MLS), and pleomorphic liposarcoma (PLS). A common characteristic of WDLS, DDLS, and PLS is amplifications in HMGA2, MDM2, and CDK4. PLS bears additional CCND1 amplifications. MLS, on the other hand, harbors a fusion of TLS/FUS–CHOP, which is responsible for pathogenesis. Angiosarcomas are diverse malignancies and bear aberrations in MYC, VEGF/VEGFR, PTPRB, and PLCG1. Leiomyosarcomas have frequent X-chromosome (Chr) gains, constitutively activated Akt and losses in Chr 10, which bears the PTEN gene. The two latter aberrations lead to mTOR activation via TSC2 and are instrumental in disease pathology. Rhabdomyosarcoma can be subtyped into alveolar rhabdomyosarcoma (ARMS) and embryonic rhabdomyosarcoma (ERMS). The former is associated with PAX3/7–FOXO1 fusions and cause Hippo-pathway dysregulation accompanied by bypass of cellular senescence, and the latter is distinguished by losses in Chr 11, along with gene mutations in the Ras pathway. Other pathways involved include Hedgehog, PI3K, and p53. Ewing’s sarcoma is characterized by EWS–ETS gene fusion, and this potent transcription factor induces genes associated with proliferation, apoptosis inhibition, and metabolic changes to favor biosynthesis and cell division. Synovial sarcoma (SS) is associated with SYT–SSX fusions: SYT–SSX2 for monophasic SS and SYT–SSX1/2 for biphasic SS. Arrows indicate gene transcription.
Liposarcoma
LPSs are mesenchymal-derived cancers that originate from adipose precursors, named so because of the resemblance they bear to fat cells when examined under microscopy.Citation9 These tumors are typically large and bulky, with extensions that branch off from the confines of the main tumor mass. LPS is the most common STS subtype, comprising 20% of all adult STS.Citation10 They most frequently occur in adults over age 40 years, and the 5-year survival rates range from 56% to 100% depending upon tumor histology.Citation11 Surgery is the standard of care for LPS, but recurrence is common and resistance to chemotherapeutics underscores a critical need for the identification of novel therapeutic targets.Citation12 The most frequent chromosome gains at the genomic level are in chromosome regions 1q, 12q, and 13q.Citation13
Liposarcoma: subtypes
The four recognized histological subtypes of LPS are categorized based on clinicopathological and molecular genetic characteristics: well-differentiated LPS (WDLS), dedifferen-tiated LPS (DDLS), myxoid LPS (MLS), and pleomorphic LPS (PLS).Citation10,Citation12 The most common are WDLS, DDLS,Citation14 and MLS.Citation9 Those with greatest metastatic potential include DDLS and PLS.Citation12 In the following sections, we present recent molecular discoveries in these biological subtypes of LPS.
Well-differentiated and dedifferentiated liposarcoma
WDLS and DDLS occur either in the retroperitoneal region or in the extremities.Citation12 Patients with retroperitoneal WDLS or DDLS have higher rates of local recurrence and disease-related deaths than those with extremity tumors.Citation12 WDLS and DDLS share common underlying genetic alterations, but WDLS in particular consists predominantly of mature adipocytes, along with mixtures of primitive lipoblasts and atypical stromal cells.Citation10,Citation12 In contrast, DDLS is believed to be more aggressive and more likely a metastatic progression of WDLS with poorer outcomes. DDLS develops due to the deregulation of normal adipocyte differentiation programs, and is thus characterized by a lack of mature adipocytes.Citation10 The exact genetic events that prompt this evolution are unclear.Citation12 WDLS and DDLS are best treated with surgical resection;Citation12 metastatic DDLS is commonly resistant to chemotherapy and radiation.Citation10
Myxoid liposarcoma
MLS and round-cell LPS constitute 30% of all LPS.Citation14 These subtypes frequently occur in the lower extremities,Citation14 with metastases observed commonly in the lungs, soft tissue, and bones.Citation15 Histologically, these cancers consist of round to oval mesenchymal cells, and MLS is characterized by the presence of lipoblasts, which are adipose precursors at different stages of differentiation, with distinct plexiform capillary patterns and a myxoid or mucous matrix.Citation9
Pleomorphic liposarcoma
PLS comprises 5% of all LPS, making it the rarest subtype.Citation14 Along with DDLS, PLS is also aggressive with metastatic potential and associated with increased disease-related deaths.Citation9 These high-grade tumors arise frequently in the retroperitoneumCitation9 and the lower extremities, and have a high risk of recurrence.Citation14 PLS has unusual histological featuresCitation9 similar to malignant fibrous histiocytoma, but with the presence of adipose differentiation.Citation14,Citation16
Liposarcoma: genomic landscape
Genomic characteristics of WDLS and progression into DDLS
A common characteristic of both WDLS and DDLS is the presence of a supernumerary ring (where the two arms of the chromosome are fused together) or a giant marker chromosome (where no structural parts of the chromosome can be identified) with amplifications in the chromosome region 12q13–12q15.Citation13–Citation15,Citation17–Citation19 This causes amplification of such genes as HMGA2, MDM2, and CDK4.Citation13–Citation15,Citation17–Citation19 The occurrence of both CDK4 and MDM2 amplification is associated with higher local recurrence rates (47% versus 12.5% in MDM2-exclusive amplifications).Citation20
Although WDLS is locally aggressive, it has relatively little metastatic potentialCitation12 and exhibits fewer copy number aberrations (CNAs) relative to DDLS: 5.7% in WDLS versus 21% in DDLS.Citation10,Citation21 Of the 11 chromosomes amplified in WDLSP, the most frequent is 12q13–12q15, found in 95% of cases with CDK4 amplifications, 87% with MDM2 amplifications, and 76% with HMGA2 amplifications.Citation10 Progression from WDLS to DDLS involves additional genomic alterationsCitation10 and importantly the downregulation of adipocyte differentiation programs.Citation10 Nine CNAs, termed progression-associated CNAs, which are differentially expressed between the two subtypes, could potentially have roles in the progression of WDLS to DDLS.Citation10
A major element of dedifferentiation from WDLS to DDLS is the loss or downregulation of adipogenesis.Citation10,Citation21–Citation24 Adipocyte-metabolic genes such as LIPE,Citation10,Citation21,Citation23 PLIN,Citation23 and PLIN2,Citation21 among others, are also uniquely absent in DDLS,Citation23 thus displaying a distinctive genomic landscape with global suppression of adipogenesis. Expression of genes related to apoptosis (BAX, BIRC5, SULF1), cytoskeleton arrangement and maintenance (CTNNB1, MARKS, TMP4, PLEC), Ras-related genes (RAB23, HRASLS3, RAB20), transcription factors (TLE4, FOXF2, SOX11), and cell-cycle control (MAPK1, CDC2, CCNB2) are differentially expressed between DDLS and WDLS.Citation23
Genomic characteristics of MLS
Interestingly, MLS displays very few genomic imbalances and in particular lacks high amplifications commonly observed in the other subtypes.Citation14,Citation25 MLS is characterized by the presence of a unique reciprocal translocation of bands 13q, which encodes for CHOP, and p11, which encodes for TLS/FUS on chromosomes 12 and 16, respectively.Citation9,Citation24,Citation25 The resulting translocation, t(12;16)(q13;p11), forms the fusion protein TLS/FUS–CHOP, which may play a role in adipose differentiation and inhibition of G1/S cell-cycle arrest induced by native CHOP proteins.Citation9 Amplifications of 13q, specifically 13q21–13q31 and 13q32, are also observed frequently in MLS and are associated with poor overall survival.Citation25 Telomerase reactivation is moderate in MLS (39%),Citation19 but the TERT promoter mutation C228T occurs commonly in MLS cases (74%).Citation26
Genomic characteristics of PLS
PLS is distinguished in having the most chromosome imbalances,Citation14,Citation16,Citation25 with more gains and deletions of chromosome regions than any other LPS subtype, occurring on all chromosomes.Citation25 Unlike MLS, PLS has not been associated with any translocations;Citation9,Citation27 instead, frequent CNA amplifications occur in a number of chromosome regions.Citation27 Specifically, amplification of 13q31–13q32 (frequent in PLS but not other subtypes) is associated with poor patient survival and increased tumor-related death, with a median survival of 35 months versus 78 months in those with no 13q gain.Citation25 PLS displays differentially high amplifications of CCND1 and similarly high amplification of CCND2, MYB, MDM2, GLI1, and CDK4 to DDLS.Citation28
Chemotherapeutics for LPS: eribulin and trabectedin
In 2015 and 2016, the FDA approved two chemotherapeutic agents specifically for LPS treatment: eribulin and trabectedin. Eribulin acts by inhibiting the polymerization of tubulin, preventing the formation of microtubules, and interfering with the mitotic spindle required for cell division. A Phase II clinical trial showed measurable tumor shrinkage and RECIST (response evaluation criteria in solid tumors) scores in LPS patients treated with eribulin.Citation29 About 47% of patients with DDLS treated with eribulin showed complete or partial response or stable disease.Citation29 Approximately 45% of patients with other LPS subtypes (eg, PLS and MLS) showed stable disease.Citation29 In a large Phase III multicenter clinical trial, eribulin treatment significantly extended overall survival in patients by 2 months compared to dacarbazine, a DNA cross-linking agent.Citation30 Overall survival was improved in LPS patients treated with eribulin compared to dacarbazine.Citation30
Trabectedin exerts its antitumor effect by interfering with DNA repair machinery and by causing DNA breakage and cell-cycle arrest. In 2007, a clinical trial with exclusively MLS patients showed efficacy (51% objective response with progression-free survival at 6 months in 88% of patients) of the drug in MLS, and specifically for those who carried the type I and II variants of the TLS/FUS–CHOP fusion products.Citation31 The efficacy of trabectedin specifically against type I and II variants was confirmed in xenograft models, showing that trabectedin prevented and prolonged the binding of variant types I and II to target genes PTX3 and NF1.Citation32
Liposarcoma: targeted therapeutics
Despite the relatively large amount of genomic information garnered for LPS subtypes, there is currently no approved targeted therapy for LPS.
CDK4/6 for WDLS/DDLS
WDLS and DDLS both harbor CDK4 amplifications,Citation13,Citation14,Citation15,Citation17–Citation19 making CDK4 a promising therapeutic target. A Phase II WDLS/DDLS-specific clinical trial showed PD0332991, a CDK4 oral inhibitor, to be effective in the treatment of these subtypes, with 60% of patients showing no disease progression at 12 weeks and median progression-free survival of 17.9 weeks.Citation33 These initial findings of CDK4 inhibition as a potential targeted therapy for LPS are promising, and several clinical trials are ongoing in patients with WDLS/DDLS.Citation34,Citation35 Furthermore, PD0332991, also known as palbociclib, has been approved for the treatment of ER-positive and human EGFR2-negative breast cancer. Given that it is the first CDK inhibitor to be approved as a cancer therapy by the FDA, its use in WDLS/DDLS could become a reality in the near future.
VEGFR/PDGFR inhibition
Treatment with pazopanib, a small-molecule inhibitor of VEGFR and PDGFR, showed promising results for patients with high–intermediate-grade disease that was surgically unresectable or metastatic.Citation36 Overall, 2.4% of patients had a partial response to the drug, 41.5% had stable disease, and 43.9% experienced disease progression; overall and progression-free survival were, respectively, 12.62 and 4.44 months.Citation36 A similar trial with pazopanib is ongoing that includes low-grade subtypes, such as WDLS.Citation37 Another clinical trial for all subtypes of LPS is investigating the efficacy of pazopanib in combination with topotecan, a compound that prevents the religation of topoisomerase-dependent DNA-strand breaks during DNA replication.Citation38
Akt–mTOR pathway inhibition
Ridaforolimus, an mTOR inhibitor, influences effector proteins S6K and 4E-BP1, and has shown promising results for several sarcomas, including LPS.Citation39 In a Phase II clinical trial, ridaforolimus was shown to elicit responses in 30% of LPS patients; 27% had progression-free survival at 6 months and median progression-free survival of 14.3 weeks, which is comparable to outcomes from other novel agents, such as trabectedin and pazopanib.Citation39 These promising findings led to a Phase III clinical trial.Citation39 An ongoing clinical trial is investigating the potential use of another mTOR inhibitor, everolimus, along with a CDK4/6 inhibitor, ribociclib, in patients with advanced DDLS.Citation40
Liposarcoma: immunotherapy
DDLS and PLS
Currently, two immunotherapeutic drug candidates are in clinical trials for these two high-grade and aggressive subtypes of LPS.Citation41,Citation42 Both trials involve monoclonal antibodies against the immunotargets PDL1 and CTLA4, namely nivolumab and ipilimumab.Citation41,Citation42 One trial is investigating the potential neoadjuvant effect of these two drugs in patients with surgically resectable tumors.Citation41 The other trial will examine the efficacy of nivolumab with or without ipilimumab in patients with unresectable or metastatic disease.Citation42
MLS
A pilot study of CAR T-cell therapy is ongoing for MLS patients with recurrent or unresectable disease. Treatment consists of administering the patients’ own dendritic cells genetically engineered to express antigen NY-ESO1.Citation43 Similarly, a Phase II clinical trial is under way looking at the effectiveness of atezolizumab, an anti-PDL1 antibody, combined with CMB305, a dendritic-cell-targeted lentiviral vector containing the NY-ESO1 sequence.Citation44 Another ongoing pilot study specific for MLS is investigating whether systemic administration of IFNγ can modulate immune-cell infiltration and expression of class II MHC proteins that are expressed on dendritic cells, phagocytes, and antibody-producing B cells.Citation45
All liposarcoma subtypes
Ongoing clinical trials are inclusively testing all LPS subtypes as well, such as an efficacy trial of the combination of trabectedin and avelumab, an anti-PDL1 antibody, in patients with unresectable and/or metastatic disease.Citation46 Another study is a Phase I trial of ABI009, an albumin-bound rapamycin compound, along with nivolumab.Citation47 A third trial is a noninferiority study looking into the effectiveness of combining the standard of care, doxorubicin, with dexrazoxane, a topoisomerase II inhibitor, and olaratumab, an anti-PDGFRA antibody.Citation48
Angiosarcoma
Making up 2% of all STS, angiosarcomas are malignant tumors that develop in the inner lining of blood vessels and lymphatic tissue. They can be found in almost any part of the body, but occur most frequently in the skin, breast, liver, and spleen.Citation49 There are several syndromes (neurofibromatosis, Maffucci syndrome, and Klippel–Trénaunay syndrome) and various exogenous chemicals (vinyl chloride, thorium dioxide, arsenic, radium, and anabolic steroids) that are known risk factors for developing angiosarcoma.Citation49 Prognosis is generally poor, since most patients are not diagnosed prior to widespread metastasis. The overall 5-year survival rate is 35%,Citation49 but varies depending on the primary tumor site. Liver and heart angiosarcomas have been found to have as low as no 5-year survival, whereas breast, skin, and soft-tissue angiosarcomas tend to have 5-year survival of 51%, 43%, and 74%, respectively.Citation50 The current standard of care is surgery and chemotherapy. A better understanding of angiosarcoma genomics could lead to targeted therapies.
Angiosarcoma: subtypes
Angiosarcomas include a mild form known as epithelioid hemangioendothelioma and more aggressive forms, simply termed angiosarcomas. Epithelioid angiosarcoma is rare and arises in endothelial cells. Angiosarcoma can be divided into five subgroups related to etiology or anatomic location: soft-tissue angiosarcoma (25%), lymphedema-associated angiosarcoma, radiation-induced angiosarcoma, primary breast angiosarcoma (8%), and cutaneous angiosarcoma (60%).Citation49 As these names imply, angiosarcoma can arise either as de novo tumors (primary angiosarcoma) or as secondary angiosarcomas due to chronic lymphedema or radiotherapy.Citation49 Because of this, women with breast cancer who undergo radiation treatment are at 1,000-fold higher risk of developing secondary angiosarcoma,Citation51 making the breast the most common place where radiation and lymphedema-associated sarcomas form.Citation51,Citation52 Furthermore, breast angiosarcoma is more aggressive than many other breast cancers and has the tendency to develop rapidly, making it difficult to treat. Although breast angiosarcoma affects deep soft tissue, it does not typically spread to the muscles of the chest wall. Other common primary angiosarcoma tumor sites include the skin and soft tissue,Citation51 with the skin of the scalp in elderly patients being the most common site for primary angiosarcoma.Citation49,Citation53 Skin angiosarcoma remains refractory to treatment measures and progresses quite rapidly. Soft-tissue angiosarcoma afflicts women and men of all ages equally, and typically presents either as a mass in the affected area or as compression of structures inside the abdomen. Although organ-specific angiosarcomas, such as those of the lungs and heart, share the same fundamental characteristics as other STS, the therapeutic strategy employed is individualized based on the subtype.
Angiosarcoma: genomic landscape
Angiosarcomas are a heterogeneous group of malignancies harboring a wide range of genetic alterations. Although critical in the pathogenesis of many cancers and even in other sarcoma subtypes like LMS and undifferentiated PLS, the role of TP53 gene alterations in angiosarcoma may be more limited. TP53 mutation and deletion rates have been shown to be as low as 4% and 0%, respectively, in angiosarcoma,Citation54 although other studies indicate higher rates.Citation52 Additionally, while mutations reported in the MAPK pathway could serve as potential targets of therapeutic interest,Citation52 this review focuses on MYC, VEGF/VEGFR, PTPRB, and PLCG1.
MYC
MYC is a proto-oncogene known to play a key role in cell-cycle progression and cell proliferation, differentiation, and apoptosis. Mutated or constitutively activated Myc has been implicated in many human cancers. For angiosarcoma in particular, MYC also plays a key oncogenic role, and the most common alterations are amplifications on chromosome 8q24.21 (50%), followed by 10p12.33 (33%) and 5q35.3 (11%).Citation55 Furthermore, MYC gene amplification and protein overexpression in angiosarcoma is well documentedCitation52,Citation55–Citation61 and is a useful tool in differentiating between primary and secondary angiosarcomas and atypical vascular lesions, which are potential precursors to angiosarcoma. Of the three most common alterations, MYC amplification has been widely shown to occur almost exclusively in secondary angiosarcoma, underscoring the fact that genetic distinction can exist even in morphologically indistinguishable tumors.Citation55,Citation57–Citation60 MYC amplification in secondary radiation-induced angiosarcoma can be observed in up to 100% of samples, as it is an early but often necessary event,Citation58–Citation61 whereas atypical vascular lesions have rarer MYC amplifications.Citation58–Citation61 Furthermore, MYC gene amplification is typically,Citation59 but not always, related to Myc protein overexpression, suggesting an alternative potential regulatory pathway of MYC expression, such as epigenetic control.Citation56 Regardless, the amplification of MYC in many cases of angiosarcoma suggests the importance of its role in the pathogenesis of angiosarcoma and its utility as a diagnostic tool, as well as a potential treatment target utilizing recently described BET inhibitors, among other agents.Citation62
VEGFR
VEGF and its receptor VEGFR play important roles in the angiogenesis of tumor tissue. VEGF overexpression has been shown in 21%–25% of STS patients of various subtypes.Citation63,Citation64 Although high VEGF expression correlates significantly with increased tumor grade in various sarcomas,Citation65,Citation66 increased VEGF serum levels are related to worse prognoses, particularly in LMS patients,Citation63,Citation64 although the use of VEGF as an independent predictor of clinical outcome as a whole remains controversial.Citation66,Citation67 Itakura et al examined immunohistochemical staining of VEGF-related proteins in 34 angiosarcoma samples and found positive expression of VEGFA (94%), VEGFC (12%), VEGFR1 (94%), VEGFR2 (65%), and VEGFR3 (79%).Citation53 Similarly, Antonescu et al showed that in 42 angiosarcoma tumor samples, 60% expressed VEGFR2 in over 75% of cells, though no CNAs were detected despite this strong protein expression and clear upregulation at the transcriptional level.Citation68 Moreover, Amo et al showed that treating an angiosarcoma cell line (ISO-HAS) with forced expression of VEGF, VEGFR1, and VEGFR2 with recombinant VEGF caused cell growth, further suggesting the importance of the VEGF family in the pathogenesis of angiosarcoma.Citation69 Targeting these VEGF-related proteins in angiosarcoma could thus prove to be an effective treatment.
PTPRB and PLCG1
Given that aberrant angiogenesis is thought to drive angiosarcoma carcinogenesis, the underlying mutational profile does in fact identify angiogenesis genes PTPRB and PLCG1 in angiosarcoma samples (n=39) examined via unbiased next-generation sequencing.Citation70,Citation71 Enrichment of both mutations was highly significant, and Behjati et al showed that 15 of 39 (38%) tumors had at least one driver mutation in signaling genes involved in angiogenesis.Citation70,Citation71 More specifically, 10 of 39 (26%) samples had inactivating PTPRB mutations, whereas 3 of 34 (9%) samples likely had activating PLCG1 mutations.Citation70 PTPRB is a tyrosine phosphatase that inhibits angiogenesis by negatively regulating VGF tyrosine kinases, including VEGFR2, and can often be truncated in angiosarcoma, which may contribute to disease pathogenesis.Citation70 In vitro models have shown that inhibition of PTPRB increases angiogenesis,Citation71 so these inactivating PTPRB mutations would be expected to lead to angiogenesis in angiosarcoma, as would the activating PLCG1 mutations. In contrast, PLCG1 encodes for PLCγ1, which is a signal transducer of tyrosine kinases. A missense alteration (R707Q) can lead to activation of this enzyme, which has been found in the autoinhibitory cSH2 domain of the protein.Citation70 This is consistent with the idea that overactive PLCγ1 drives angiosarcoma by constitutive signal transduction downstream of receptor tyrosine kinases, reinforcing PLCG1 as an attractive therapeutic target in angiosarcoma.Citation70 In addition to these two genes, other rare mutated genes, such as PIK3CA (of 39 cases), FLT4 (1 of 39 cases) and H/K/NRAS (5 of 39 cases) have also been reported in angiosarcoma.Citation70
Angiosarcoma: targeted therapeutics
Tyrosine kinase inhibitors
Cytotoxic chemotherapy drugs like ifosfamide, anthracyclines, and taxanes are currently used in treating angiosarcoma.Citation68,Citation72 However, recently developed anticancer drugs that target angiogenesis-related proteins are exciting, because of the role of the VEGF family in the pathogenesis of this family of diseases. Overexpressed angiogenesis-related proteins have recently been targeted with tyrosine-kinase inhibitors that inhibit proteins in the VEGF family with the hope that curtailing the blood supply might cause tumor shrinkage. These inhibitors include sunitinib, sorafenib, and pazopanib, which target VEGFR1, VEGFR2, and VEGFR3.Citation68
In COS7 cells transfected with two different activating mutant forms of VEGFR2, the tyrosine kinase inhibitors sunitinib and sorafenib were effective in decreasing autophosphorylation of both mutants, suggesting their potential for use in angiosarcoma patients bearing mutant VEGFR2. This mutation has been reported in 10% of samples.Citation68 In a recent Phase II study, von Mehren et al showed that sorafenib treatment led to a progression-free rate of 38% in the vascular sarcoma cohort (63% of which were angiosarcoma patients).Citation73 Additionally, the effectiveness of sorafenib in angiosarcoma appears to be related to baseline circulating VEGFA levels, with lower levels being significantly correlated with better outcomes.Citation74
The first Phase I dose escalation study of pazopanib in various advanced cancers showed that it was generally well tolerated and had an antitumor activity in a variety of cancers.Citation75 Since then, other clinical trials have confirmed both the safety and the efficacy of pazopanib in STS,Citation76 establishing it as a viable new treatment option for select STS patients.Citation77 Pazopanib proved particularly effective in patients with elevated levels of VEGFR2 in their tumors, as their median overall survival was 7.2 months compared to 2.3 months in those patients with low expression,Citation78 further highlighting the value of targeting VEGFR2 overexpression in angiosarcoma as a treatment option.
Anti-VEGF antibody
Use of the anti-VEGF antibody bevacizumab in angiosarcoma has shown promising results alone and in combination with traditional treatments like surgery, chemotherapy, and radiotherapy. Rosen et al showed that bevacizumab monotherapy in one patient with facial cutaneous angiosarcoma who was unable to undergo traditional treatment showed a well-tolerated and encouraging partial response.Citation79 Similarly, a Phase II study of bevacizumab use in angiosarcoma (23 of 30) and epithelioid hemangioendothelioma (7 of 30) patients reported that 9% of angiosarcoma patients showed a partial response, and 48% of angiosarcoma patients had stable disease with an average time to progression of 26 weeks. Bevacizumab was also well tolerated by these patients.Citation80 Furthermore, use of bevacizumab in combination with preoperative radiotherapy followed by resection of the tumor bed in two cases of angiosarcoma of the nose was very effective: both patients had a complete response, no residual disease, and no recurrence after follow-up of 8.5 months and 2.1 years.Citation81 Fuller et al showed that a combination of bevacizumab with chemotherapy was also effective in even inoperable angiosarcoma, with one patient showing dramatic improvements in appearance and symptoms, which remained stable 11 months after treatment had ended.Citation82 Finally, in a Phase II trial of a combination of gemcitabine, docetaxel, and bevacizumab in various STS types, 60% of angiosarcoma patients showed a partial response to this very exciting group of anticancer drugs.Citation83
Angiosarcoma: immunotherapy
Recently, the advent of immunotherapy has provided promising alternatives to cytotoxic chemotherapeutic agents and targeted therapies in cancer treatment. The most popular immune system targets have been the checkpoint proteins CTLA4 and PD1/PDL1, which can modulate the immune system in the control of cancer progression. Unfortunately, ipilimumab, an anti-CTLA4 antibody, showed no response in six patients with SS.Citation84 Immunotargeting of the PD1 receptor and its ligand PDL1 has become increasingly popular in the treatment of multiple cancers, including non-small-cell lung cancer, melanoma, and renal and bladder cancers, and reports of responses to these agents in patients with angiosarcoma have begun to emerge.Citation85
Leiomyosarcoma
LMS tumors that originate from smooth muscle connective tissue account for 10% of all soft-tissue sarcoma.Citation86 LMS frequently occurs in the extremities, small intestine, or retroperitoneal spaces, or most commonly in the uterus,Citation87 hence the categorization into either uterine LMS (ULMS) or nonuterine LMS (NULMS).Citation86,Citation88 ULMS, which accounts for 1% of all uterine malignanciesCitation89 and 40% of all uterine sarcomas,Citation86 is highly aggressive, with greater metastatic potential than NULMS;Citation86 it is also resistant to chemotherapy and radiotherapy.Citation90 Median survival in NULMS and ULMS is about 8 and 4.2 years, respectively.Citation86 ULMS can progress de novoCitation89 or as a result of transformation from uterine leiomyoma, smooth muscle hyperplasia that occurs in as many as 80% of women.Citation91 The documented incidence of transformation from uterine leiomyoma to ULMS, however, is rare (<0.1%).Citation89 The 5-year survival rate for LMS is 40%, but decreases to 10%–15% for high-grade LMSCitation92 and 15%–25% for ULMS.Citation90,Citation91 The standard of care for LMS is surgical resection when possible.Citation86 LMS can remain dormant for extended periods, and the best outcomes occur after early surgical excision with wide margins.Citation92,Citation93 The 5-year rate of relapse is 40%, which is associated with very high mortality.Citation92,Citation93
Leiomyosarcoma: subtypes
Guo et al confirmed the presence of three molecular subtypes of LMS.Citation87 Types I and II are linked with extrauterine sites, and type III is closely associated with ULMS. Type I can be identified through immunostaining of overexpressed markers, such as ACTG2, SLMAP, LMOD1, CFL2, and MYLK. Type II is characterized by overexpression of ARL4C, associated with translation, translational elongation, and protein localization, and overexpression of CDK4, CTNNB1, AURKA, RHEB, EGFR, CCND1, MTOR, MAPK1, NOTCH2, and ROR2 has been reported. Type III is associated with upregulation of pathways involved in metabolic processes, ion transport, and regulation of transcription; overexpressed genes include MDM4, ERBB3, EPHA3, ESR1, and EGFR. The identification of these molecular subtypes has been fairly recent, and although more investigations are warranted, these differences could have clinical significance related to the use of existing or novel targeted therapies.
Leiomyosarcoma: genomic landscape
Genomic imbalances are observed in 88% of LMS cases,Citation94 and 60% of them have aberrant chromosome numbers and structures. More aberrations are reported in higher grade tumors than in lower grade ones.Citation95 LMS tumors have pleomorphic histology,Citation88 absence of CD44 variant 3,Citation96 CD34, c-Kit, and S100 expression,Citation94 and complex karyotypes.Citation88 Almost half of ULMS cases (48%) have X-chromosome gains, and the associated amplicons are located near regions containing the androgen receptor, which might potentially contribute to ULMS resistance to hormone therapy.Citation90 Other associations include the putative oncogenes ELK1 and ARAF1.Citation95 Sixty-two percent of ULMS cases display loss of the chromosome region 10q, which harbors the tumor suppressors PTEN and MXI1, and loss of 10q is associated with recurrentCitation95 and higher grade tumors.Citation94,Citation95 Most ULMS cases show loss of 13q, the region that houses the tumor suppressor RB.Citation95 Loss of 13q, however, is associated with better prognosis than loss of 10q and contributes to the early development of LMS.Citation95 Promoter hypermethylation of the tumor suppressor RASSF1A occurs in 39% of LMS, which is higher than in other sarcomas, such as LPS and malignant fibrous histiocytoma, and is associated with poor prognosis in LMS patients with stage II and III cancers.Citation93
PTEN–Akt–mTOR pathway in leiomyosarcoma
Most LMS cases are reported to have activated Akt,Citation88 and as mentioned earlier, loss of 10q, which contains the PTEN tumor suppressor gene, is a frequent genomic abnormality found in ULMS and associated with recurrenceCitation95 and high-grade tumors.Citation94,Citation95 Hyperplastic smooth muscle cells that lose PTEN expression then show constitutive activation of Akt, which results in malignant progression into LMS through the release and activation of mTOR via TSC2.Citation88 Mice deficient in PTEN in smooth muscle lineage cells have shown decreased life spans, with widespread smooth muscle hyperplasia, mainly in the blood vessels, and urinary and intestinal tracts as early as 1 month.Citation88 This was accompanied by rapid onset and an 80% increase in incidence of LMS as early as 2 months after birth.Citation88 Interestingly, there was no appearance of ULMS, suggesting an altered molecular pathogenesis.Citation88 When these 1-month-old mice were treated with an mTOR inhibitor, rapamycin, there was a significant increase in life span and a decrease in tumor growth accompanied by decreases in pAkt and mTOR target pS6, emphasizing the crucial role of this pathway in LMS tumorigenesis.Citation88
Leiomyosarcoma: targeted therapeutics
In addition to its use in LPS, the chemotherapeutic agent trabectedin has been approved by the FDA specifically for the treatment of LMS. However, since more than half of early-stage patients experience relapse after therapy,Citation97 and the ULMS subtype is resistant to chemotherapy,Citation90 more targeted therapies are required for the treatment of LMS.
Aurora kinase A inhibition
Proteins that regulate the formation of the mitotic spindle during cell division are frequently overexpressed in a number of cancers.Citation98 One of the key players in mitotic spindle organization and stability is aurora kinase A (AurKA), and its expression is highly regulated in ULMS compared with benign LMM or normal myometrial tissue.Citation91 Shan et al illustrated the efficacy of an AurKA inhibitor, MK5108, in a mouse model of LMS in which treatment decreased tumor growth and induced G2/M cell-cycle arrest and apoptosis.Citation91 A Phase I clinical trial for the use of MK5108 in solid cancers indicated that it was well toleratedCitation91 and provides the impetus for testing this inhibitor in LMS and other sarcomas.
Combination of aurora A kinase and mTOR inhibition
As mentioned previously, activation of the Akt–mTOR pathway through the loss of tumor suppressor PTEN is crucial for the development of LMS.Citation88,Citation94,Citation95 One group is investigating the potential therapeutic benefits of simultaneous AurKA and mTOR inhibitionCitation97 by using the AurKA inhibitor MLN8237, along with the mTOR inhibitor rapamycin. They found that with a specific schedule of 24 hours of pretreatment with MLN8237 followed by cotreatment for 72 hours with both MLN8237 and rapamycin, tumor volume decreased significantly when compared to MLN8237 or rapamycin alone; this was accompanied by a pronounced decrease in cell proliferation and an increase in apoptosis.Citation97 Interestingly, LMS subtype II shows overexpression of both MTOR and AURKA, making the dual AurKA and mTOR inhibition regimen a possible personalized therapy for patients with this particular subtype.
ROR2 as potential therapeutic target in leiomyosarcoma
ROR2 is activated by Wnt5A via the noncanonical Wnt pathway and is highly expressed in several sarcoma subtypes, including LMS.Citation99 LMS patients with strong ROR2 staining have worse 5-year disease-specific survival than those with weak or undetectable ROR2 staining.Citation99 It is noteworthy that ROR2 expression is consistent between primary and metastatic LMS tumors, which might enable a common treatment for both.Citation99 Edris et al showed that ROR2 knockdown led to a 50% decrease in invasiveness of LMS cell lines and a threefold reduction in average xenograft tumor mass in mice.Citation99 ROR2 expression is specifically enriched in subtype II, making ROR2 another potential therapeutic target for this subtype. Therefore, the use of Wnt pathway inhibitors, such as OTSA101, an anti-Fzd10 antibody in a Phase I trial for advanced SS treatment, might prove to be effective in LMS.Citation100
EGFR inhibition
Using testicular LMS as a model, Sette et al uncovered a putative LMS cancer stem-cell population that was resistant to chemotherapy.Citation101 Specifically, this population and differentiated tumor populations have shown high activation of EGFR.Citation101 High EGFR expression is also observed in LMS specimens compared to normal tissue. EGFR inhibition combined with chemotherapy results in a decrease in tumor size accompanied by an increase in apoptosis.Citation101 As such, EGFR inhibitors could be used to target both cancer stem cells and differentiated tumor populations in LMS. This therapeutic modality can potentially be effective for those with subtype III, who have enriched EGFR expression.
LMP2 as a potential target for ULMS
PSMB9/LMP2 encodes a subunit of the proteasome that is involved in antigen processing and frequently genetically altered in ULMS.Citation90 LMP2 loss is observed in 85% of ULMS cases,Citation90 and over 30% of samples have essential mutations in LMP2.Citation90 Female mice with mutated copies of LMP2 spontaneously develop ULMS by 14 months, with 40% prevalence.Citation102 One group showed that inoculation of ULMS cells expressing exogenous LMP2 led to a reduction in tumor growth with no toxicity.Citation90 Unfortunately, the proteasome inhibitor bortezomib alone showed the minimal activity in a Phase II study of STS, but one of the patients studied did have a partial response, indicating that combination therapy with other agents may have better effects.Citation103 While surgery is a treatment option for resectable ULMS, targeting and reactivation of LMP2 could potentially be efficacious for this category of LMS.
Hepatocyte growth factor/scatter factor
When hepatocyte growth factor/scatter factor (HGF/SF) is bound to its receptor, c-Met, it promotes angiogenesis, proliferation, and invasion of cancer cells.Citation104 Since the c-Met protein is overexpressed in LMS,Citation104 it may be a prime target amenable to therapeutic intervention. Burgess et al developed a fully human anti-HGF/SF antibody termed AMG102/rilotumumab,Citation105 which significantly decreased tumor growth when used to treat LMS in tumor-bearing mice.Citation104 There have been several completed clinical trials for AMG102 in other nonsarcoma cancers;Citation106–Citation110 one, in particular, showed promising results when combined with epirubicin, cisplatin, and capecitabine for gastric and esophagogastric cancers.Citation106 Another clinical trial is ongoing for squamous-cell lung carcinoma,Citation111 but no study has yet investigated the efficacy of AMG102 in either LMS or other sarcomas. Based upon the encouraging in vivo mouse work and the promising human clinical trials with other types of malignancies, there is strong rationale for testing AMG102 in LMS and other sarcomas.
Leiomyosarcoma: immunotherapy
Checkpoint inhibition
Pembrolizumab, an anti-PDL1 antibody, was approved by the FDA in 2017 for solid tumors.Citation112 A number of LMS tumors are positive for PD1 expression, and two clinical trials are investigating the effect of pembrolizumab aloneCitation113 or in combination with the immunosuppressor cyclophosphamideCitation114 in LMS. A different anti-PDL1 antibody, nivolumab, was shown to be an effective treatment for one patient with refractory LMS, who had already undergone surgery and multiple rounds of radiation and chemotherapy.Citation115 Furthermore, Paoluzzi et al reported a retrospective study of 24 STS patients treated with nivolumab: seven were diagnosed with LMS, three of whom had stable disease after eight cycles of treatment.Citation112 However, the response to pembrolizumab was dramatically different in different individual tumors in a single ULMS patient.Citation116 Molecular analysis suggested that loss of PTEN in LMS may correspond to resistance to PD1 inhibition.Citation116 Since PTEN loss or its equivalence is a frequent genetic alteration in LMS,Citation88,Citation94,Citation95 other immunotherapeutic strategies may be considered for patients with these tumors. Overall, immunocheckpoint inhibitors have shown encouraging activity in LMS patients, and studies with expanded cohort enrollment are ongoing to confirm the efficacy of these inhibitors.
CD47 inhibition
CD47 is a cell surface marker that is overexpressed by cancer cells (87% of cases) relative to normal muscle tissues; CD47 prevents cells from being phagocytized by macrophages of the immune system.Citation92,Citation117 LMS has also shown high infiltration of tumor-associated macrophages, which can promote cancer-cell aggressiveness, and patients have shown significantly poorer prognosesCitation92,Citation117 Edris et al hypothesized that blocking the antiphagocytosis function of CD47 would allow infiltrated macrophages already present in the LMS tumor to switch from a protumor to antitumor function and eliminate the tumor cells within the tumor.Citation92,Citation117 In vitro cell-based studies demonstrated phagocytosis of LMS cells by macrophages when treated with an anti-CD47 antibody.Citation92 Anti-CD47 treatment drastically reduced tumor mass in vivo in the range of 5- to 30-fold, with few to no distal metastases compared to the controls; a nearly 70-fold decrease in distal metastases was also reported.Citation92 Currently, four different anti-CD47 antibodies are undergoing clinical trials for use in hematological and solid cancers.Citation118–Citation123 The aforementioned studies collectively represent an underexploited therapeutic opportunity for treatment of LMS patients with anti-CD47 antibodies.
Rhabdomyosarcoma
RMSs are highly aggressive tumors that typically develop from skeletal muscle cells.Citation124 They represent 3%–4% of all childhood cancers and are the most common childhood and adolescent STS,Citation124,Citation125 accounting for 40% of pediatric STS.Citation126 Although RMS can occur anywhere in the body, it most commonly occurs in the head and neck (10%), orbit (9%), genitourinary tract (24%), extremities (19%), and nasal passage and sinuses (16%).Citation124,Citation127 Not only are RMS symptoms tumor site-specific, but prognoses are also linked to primary tumor location. Standard of care depends on primary tumor site and the age of the patient, but can include surgery, radiation, and chemotherapy. RMS frequently metastasizes to the lungs, bone marrow, and bones, and heterogeneity in these tumors makes them confounding and difficult to diagnose, given the lack of strong genetic markers. However, up to 70% of newly diagnosed cases that do not involve metastases can be cured with multimodal therapy. Since survival rates can vary between 35% and 90% depending on the RMS subtype, a clear diagnosis is essential for disease management.Citation128,Citation129 Various environmental risk factors have been associated with increased risk of developing RMS, such as paternal smoking, maternal recreational drug use, advanced maternal age, and X-ray exposure in utero.Citation126 Additional genetic risk factors include neurofibromatosis type 1, Li–Fraumeni syndrome, Beckwith–Wiedemann syndrome, hereditary retinoblastoma, nevoid basal-cell carcinoma syndrome, Rubinstein–Taybi syndrome, and Costello syndrome.Citation124,Citation126 Though advancements in multimodal chemotherapy have shown large increases in patient survival,Citation130,Citation131 toxicity remains an issue, and the 5-year survival rate for metastatic disease remains at 30%,Citation132 underscoring the need for additional therapeutic strategies.
Rhabdomyosarcoma: subtypes
RMS has traditionally been categorized into two main types according to histopathological differences. The most common subtype is embryonic RMS (ERMS), which accounts for about 60% of RMS,Citation133 whereas alveolar RMS (ARMS) accounts for ~20% of cases.Citation133 ERMS usually manifests in the head and neck, genitourinary tract, and retroperitoneum of children <10 years of age, whereas ARMS usually occurs in the trunk, arms, and legs of adolescents and young adults.Citation124 Clinical outcomes differentiate the two subtypes as well, because outcomes for ERMS are typically considered favorable if the tumor is localized, whereas ARMS has a higher propensity to metastasize and generally has a poorer prognosis.Citation124,Citation132 Five-year survival rates for RMS vary depending on the risk group and subtype,Citation130 but the overall ERMS 5-year survival rate is 73.4%, whereas the ARMS 5-year survival rate is 47.8%.Citation126
Rhabdomyosarcoma: genomic landscape
In RMS, the overall somatic mutation burden is relatively low,Citation132 but various chromosomal alterations are key and could serve either as prognostic indicators or as targets for therapy.
Alveolar rhabdomyosarcoma
Eighty percent of ARMS have distinguishing translocations between chromosomes 2 and 13 (t[2;13][q35;q14]) or between chromosomes 1 and 13 (t[1;13][p36;q14]), which correspond, respectively, to PAX3–FOXO1 and PAX7–FOXO1 gene fusions.Citation124,Citation132–Citation134 PAX3–FOXO1 gene fusions have been detected in 55% of ARMS patients, whereas PAX7–FOXO1 gene fusions were found in 22% of ARMS patients.Citation132 Although PAX3, PAX7, and FOXO1 are typically transcription factor-encoding genes, the PAX3–FOXO1 fusion gene produces an even more potent transcription activator than PAX3, suggesting a role in the pathogenesis of ARMS through aberrant upregulation of PAX3 target genes,Citation135,Citation136 though PAX3–FOXO1-specific targets, such as PDGFR, have also been shown to be upregulated.Citation137 In vitro and in vivo experiments have confirmed that PAX3 and PAX3–FOXO1 compete for the same targets, and higher PAX3–FOXO1 embryonic expression leads to impaired neural crest migration and development.Citation136 Although PAX3–FOXO1 expression seemingly plays a critical role in pathogenesis, it alone does not seem to be enough to cause ARMS. Mouse models made to express the gene fusion developed physical abnormalities but no tumors,Citation135,Citation138 suggesting the need for coexisting alterations. Potential cooperating events seem to include dysregulation of the Hippo pathwayCitation139 and PAX3–FOXO1 bypassing cellular senescence by cooperating with loss of INK4a.Citation140 Whole-genome sequencing suggests that the most common cooperating events in PAX3/7–FOXO1 fusions present in RMS are due to genetic amplifications of MYCN, CDK4, and MIR17-92; deletion of CDKN2A; or loss of heterozygosity of chromosome 11p15.5.Citation124,Citation132
Whereas the PAX3/7 fusion type was not associated with patient outcome among ARMS patients with localized disease, patients with metastatic ARMS with PAX7–FOXO1 fusion had a 4-year survival rate of 75% compared to an 8% 4-year survival rate for those with the PAX3–FOXO1 gene fusion.Citation134 The presence or absence of these PAX3/7–FOXO1 fusions has been used to subcategorize RMS more accurately as a whole,Citation132,Citation141 since the prognosis and molecular profiling for fusion-absent ARMS patients are nearly indistinguishable from those of ERMS patients, despite their histological differences.Citation141 ARMS samples that were fusion absent also had significantly more somatic mutations than those that were fusion present,Citation132 suggesting the need and possibility for alternative treatment strategies between the two. Mutations found in fusion-absent but not fusion-present ARMS include the genes NRAS and PIK3CA, whereas fusion-positive tumors nearly exclusively showed amplification of the chromosome region 12q13–12q14,Citation132 which is associated with worse overall survival.Citation142
Embryonic rhabdomyosarcoma
ERMS most characteristically shows allelic loss at chromosome 11p.15.5,Citation124 a region that appears to include tumor-suppressor genes, and wild-type chromosome 11 transfer into an ERMS cell line causes a decrease in proliferation,Citation143 suggesting restoration of tumor suppressor activity. Studies suggest that in most ERMS, both 11p.15.5 alleles are inactivated, with an inactivated paternal allele being conserved and the maternal allele being lost altogether.Citation124 Further chromosomal alterations shown in ERMS include gains of chromosomes 2, 7, 8, 11, 12, 13, 17, 18, and 20; losses of chromosomes 10, 14, 15, and 16;Citation144,Citation145 and translocations in the 1p11–1q11 region.Citation146 Moreover, studies have shown that 35% (5 of 14) of ERMS samples contain mutant NRAS or KRAS genes,Citation147 and at least 45% of fusion-absent RMS as a whole have mutational activations in the Ras pathway, including FGFR4, RAS, NF1, and PIK3CA.Citation132 Additional mutations or gene amplifications have also been shown in TP53, MDM2, CDKN2A, GLI1, CTNNB1, and PTPN11.Citation124,Citation132,Citation148–Citation150 Though p53 expression has been shown in ample RMS,Citation151 P53 genetic alteration frequencies in RMS seem to vary. Takahashi et al reported P53 gene alterations in 22.2% (10 of 45) of samples,Citation151 but Taylor et al reported that only 5% (1 of 20) of tumor samples showed P53 mutations.Citation152 Despite these differences, neither source reported a correlation between P53 mutation status and prognosis.Citation127,Citation152 Both sources also reported similar MDM2 gene amplification frequency in RMS samples, with Takahashi et al reporting amplification in 16.7% (3 of 18) of samplesCitation127 and Taylor et al reporting 10% (2 of 20).Citation152 Additionally, Paulson et al showed CDKN2A/B focal deletion in 23% (6 of 26) of ERMS, activating FGFR4 mutations in 20% (5 of 26) of ERMS, frequent low-level gains of a chromosome region containing GLI1, deletions in the NF1 locus in 15% (4 of 26) of ERMS, and RAS-family activating mutations in 42% (11 of 26) of ERMS.Citation149 Similarly, Pressey et al showed that high expression of GLI1 was present in 21% (15 of 70) of ERMS tumors,Citation148 and Shukla et al showed RAS family mutations in 11.7% (7 of 60) of ERMS samples, FGFR4 mutations in 9.3% of ERMS samples, and PIK3CA mutations in 4.9% of ERMS samples.Citation150 Collectively, these studies suggest that p53, RAS, Hedgehog, and PI3K pathways are potentially necessary in the pathogenesis of ERMS and could thus be sensitive to targeted inhibition.
Rhabdomyosarcoma: targeted therapeutics
Standard chemotherapy treatment from the Soft Tissue Sarcoma Committee of the Children’s Oncology Group consists of stratification of patients based on risk groups and undergoing regimens with combinations of vincristine, dactinomycin, cyclophosphamide, and sometimes irinotecan.Citation131 Although success has been reported with low-risk and intermediate-risk groups, the 5-year failure-free survival for high-risk groups has changed little over the past 25 years.Citation130,Citation133 While a greater understanding of the molecular basis of RMS has led to new strategies of stratifying patients into more accurate risk groups to improve chemotherapeutic outcomes,Citation133,Citation153 alternative targeted therapies also show promise. One study showed RMS cell lines were sensitive to an IGF1 receptor small-molecule inhibitor.Citation154 Similarly, taking advantage of inhibitor pathway activity in RMS, investigators showed in two studies that combination treatment of Ras–MEK–ERK and PI3K–Akt–mTOR pathway inhibitors led to synergistic RMS inhibition in vitro and in vivo.Citation155,Citation156 Guenther et al used the dual PI3K–mTOR inhibitor PI103 in combination with the MEK inhibitor U0126 on RMS cell lines and found highly synergistic triggering of apoptotic activity in both histological variants, whereas use of only one drug failed to cause cell death.Citation156 Renshaw et al inhibited the same pathways, but used a combination of the TORC1/2 inhibitor AZD8055 and the MEK inhibitor AZD6244, and were able to show synergistic cell growth inhibition in RMS xenografts.Citation155 Interestingly, their study also showed a lack of efficacy when just one drug was used, because compensatory cross-talk pathways seemed to render monotherapy ineffective. Finally, Chen et al showed that RMS is susceptible to reactive oxygen species and suggested that therapeutics that increase oxidative stress may synergize with current chemotherapy treatments against RMS.Citation157,Citation158
Rhabdomyosarcoma: immunotherapy
The oncogenic protein Pax3–FoxO1 plays a role in the development of RMS and promotes an immunosuppressive tumor microenvironmentCitation153 that renders antitumor function by the immune system ineffective. In the following sections are some highlights of recent breakthroughs and current immunorelated clinical trials for RMS.
Selective autologous lymphocyte and immunostimulator regimen
Although localized RMS is quite treatable, recurrent or metastatic disease is associated with disappointing outcomes.Citation159 In an early clinical trial looking into the efficacy of an adjuvant immunotherapy for recurrent or metastatic RMS, patients received infusions of their own lymphocytes with or without dendritic cells pulsed with the fusion peptide plus IL2, an immunostimulator.Citation159,Citation160 The initial results were promising, with 43% 5-year overall survival for those treated with the combination of lymphocytes, peptide-pulsed dendritic cells, and IL2 compared to 31% among those treated with lymphocytes alone.Citation159 A second-generation clinical trial, which enrolled similar participants, modified the regimen to induce a greater immunoresponse and antitumor effect.Citation161 The new protocol further enriched and purified the lymphocytes to be depleted of CD25+ regulatory T cells that caused immunosuppression, as well as potential residual tumor cells. The mature dendritic cells were pulsed with the patient’s own tumor lysate in place of the fusion peptide, and the immunostimulant added was IL7, as opposed to IL2.Citation159,Citation161 The regimen was tolerated well, with no treatment-related high-grade adverse effects.Citation159 The 5-year overall survival rate with this second-generation immunotherapy was 51%, which was significantly higher than the previous regimen, and the progression-free survival was 32%.Citation159 Interestingly, survival on this regimen was higher for RMS patients than for those with other sarcomas, suggesting the specificity of this regimen to those with metastatic or recurrent RMS.Citation159
Oncolytic viruses in the treatment of RMS
Two trials are investigating the effectiveness of two different oncolytic viruses in the treatment of RMS.Citation162,Citation163 Oncolytic viruses can specifically target dividing, cancerous cells while sparing the differentiated, normally functioning cells, and both trials are in the early phase of safety and dose escalation testing. One trial, though completed with no results posted, investigated the vaccinia virus armed with an immunostimulatory GM-CSF.Citation162 The other trial, ongoing, is examining the antitumor effect of a herpes virus.Citation163
Other current clinical trials
A clinical study of the effect of a tumor lysate vaccine plus the cytokine IL7 showed very promising initial findings, with >50% of participants showing positive immunoresponses.Citation164 Another clinical trial that specifically enrolled only RMS patients is testing the feasibility of cytotoxic T cells armed with tumor-associated antigens and investigating whether an antitumor response can be specifically launched against cancer cells.Citation165 The five antigens being tested are SSX, survivin, NY-ESO1, MAGEA4, and PRAME.Citation165 Lastly, one clinical trial is investigating the effectiveness of a drug-conjugated antibody, lorvotuzumab mertansine, in which an anti-CD56 antibody is conjugated to the drug mertansine, a tubulin inhibitor, in RMS and other sarcomas.Citation166
Ewing’s sarcoma
ES is a neuroectodermal-related malignancy of the bone and soft tissue.Citation167,Citation168 ES occurs in children and young adults,Citation169 with higher frequency in males.Citation167,Citation168 Frequent primary ES sites include the paravertebral region, the chest wall, and the lower extremities.Citation167 For patients with localized disease, the 5-year relapse-free survival rate is 50% for axial primary sites and 67% for all other sites, but decreases drastically to 21% for those with detectable metastasis at diagnosis.Citation168 However, 30%–50% of those with localized disease will experience relapse within 3 years.Citation167 In one study, surgery decreased the relapse rate significantly, from 31% to 15% for axial tumors and from 20% to 4% for other sites, but patients whose disease relapsed within 2 years typically had worse survival rates.Citation168 The hallmark of ES is the translocation fusion between the chromosome regions of the EWS–ETS family of transcription factors.Citation169,Citation170 This cancer is grouped together with primitive neuroectodermal tumors and termed the “Ewing family” of tumors due to the presence of similar EWS–ETS translocations. The difference between the two is a continuum of neural differentiation, with one end of the spectrum being primitive neuroectodermal tumor with its differentiated neural phenotype predominantly found in soft tissue, and the other being ES with its undifferentiated neural components.Citation167 The extent of differentiation is not a significant prognostic factor for patients with Ewing family tumors.Citation129,Citation171
ES: subtypes
ES tumors are tightly compacted; comprise small, rounded malignant cells separated by strands of fibrous tissue; and contain little to no intercellular stroma.Citation169 ES tumors arise from embryonic osteochondrogenic progenitors that positively express ERG, GDF5, and PTHLH.Citation172 ES and the Ewing family of tumors are characterized by the translocation of EWS and the ETS family of transcription factors, mainly in FLI1 (≥85% of cases) and ERG (10% of cases),Citation167,Citation170,Citation173 and even some rarer fusions with ETV1, E1AF, and FEVCitation169 (1%–5%).Citation173 There are four structural variants of EWS–ERG fusion transcripts and up to 18 possible variations of EWS–FLI1 transcripts, with the most common being types I and II.Citation167 Previously, type I showed significantly higher median overall survival compared to other transcript types (9 versus 2 years),Citation167 but with recent treatment advancements for ES this disparity is now equalized.Citation174 There are very few mutated genes observed in ES related to signaling pathways and chromatin-modifying genes.Citation175 Infrequently but consistently, aberrations are detected in three genes: STAG2 (15%–17%), CDKN2A (12%–22%), and TP53 (6%–7%).Citation175,Citation176 STAG2 and CDKN2A mutations are mutually exclusive and observed in primary tumors, as well as cell lines.Citation176 Patients carrying STAG2 or TP53 mutations or both have much lower survival rates.Citation176 Interestingly, while only 6% of ES patients show aberrations in TP53, this increases to 25% after treatment, suggesting the role of TP53 deregulation in treatment resistance and recurrence.Citation175
ES: genomic landscape
The translocation fusion between EWS and ETS family members produces a potent oncogenic transcription factorCitation177 capable of inducing tumorigenesis through increased cell viability and proliferation,Citation178–Citation181 apoptosis inhibition,Citation180 metabolic changes to favor biosynthesis, and subsequent cell division.Citation182 EWS–ETS regulates cell proliferation and anchorage-independent growth in ES cells, but not in a non-ES cell line.Citation178 It also regulates cell viability through LRWD1, which plays a role in stabilizing the origin recognition complex required for precise DNA replication.Citation181 Additionally, EWS–ETS induces autophagy in ES through overexpression of an autophagy-related gene, ATG4B, which leads to a higher rate of proliferation and lower rate of apoptosis.Citation180 Metabolism in ES is also altered due to EWS–ETS oncogenic regulation, which increases serine biosynthesis via PGHDH upregulation, for the production of proteins, lipids, and nucleic acids to meet the demands of cell proliferation.Citation182 Interestingly, elevated PGHDH expression is highest in ES compared to other cancer cell lines, as well as normal tissue, and patients who are deemed at high risk show upregulation in PGHDH.Citation182 Inhibition of PGHDH decreases cell proliferation in ES, but not in other non-ES cell lines, suggesting this PGHDH-dependent metabolic phenotype is found exclusively in ES.Citation182
Metastasis is a crucial factor leading to mortality in ES.Citation168 Recently, Choo et al demonstrated the importance of TWIST1, a transcription factor involved in early development, in ES metastasis.Citation183 TWIST1 silencing in an in vivo xenograft model showed decreased metastatic burden, and regardless of metastasis status in patients, positive expression of TWIST1 showed a trend toward lower survival.Citation183 Another recent discovery was the oncogenic potential of KDM3A, a histone demethylase, in ES.Citation184 KDM3A silencing in vitro showed a 50% decrease in migration and invasion and a tenfold decrease in metastatic burden compared to controls (in mice).Citation184 MCAM, which is also involved in metastasis in other cancers, is a direct downstream target of KDM3A, and when silenced, recapitulated impaired proliferation and metastasis are observed with KDM3A silencing. MCAM was also significantly associated with poor survival in patients.Citation184 ES cells with elevated expression of APLP2, a prosurvival mediator, are resistant to irradiation as well as immune-cell killing via lymphokine-activated killer cells,Citation185 allowing cells to continue to grow after treatment and metastasize.
ES: targeted therapies
Without systemic chemotherapy, most ES patients develop rapid tumor recurrence.Citation186 Standard chemotherapeutic treatment includes combinations of vincristine, actinomycin D, cyclophosphamide, doxorubicin, etoposide, and ifosfamide.Citation186,Citation187 Although the 5-year survival rate of patients with localized disease is relatively high (70%), thanks to recent advancements in diagnosis, surgery, chemotherapy, and radiation, survival rates for patients with metastatic or recurrent disease remain <25%.Citation186 Long-term toxic effects of treatment continue to be a major issue,Citation188,Citation189 further emphasizing the need for new forms of therapy.
Because of the characteristic presence of the fusion gene EWS–FLI1 in ES, it has been the target of various therapeutic attempts that have yielded promising in vitro and in vivo results. Use of a small molecule, YK4-279, to bind to EWS–FLI1, thereby inhibiting its usual binding and transcriptional modulation, caused apoptosis induction in ES cells and reduced the growth of ES orthotopic xenografts.Citation190 A recent study found synergism in the inhibition of EWS–FLI1 activity between YK4-279 use in combination with vincristine both in vitro and in vivo.Citation191 The YK4-279 analog TK216 is currently being used in a Phase I clinical trial in patients with relapsed or refractory ES.Citation192
Moreover, although trabectedin is a chemotherapeutic, trabectedin sensitivity has been shown to be specifically associated with changes in EWS–FLI1 transcription factor activity, because drug treatment decreased the expression of several downstream targets of the fusion gene.Citation193 Despite this, a Phase II clinical trial using trabectedin in patients with recurrent RMS, ES, and non-RMS soft-tissue sarcomas showed the insufficient activity of the drug as monotherapy.Citation194 By taking advantage of downstream targets, Grohar et al were able to show that trabectedin led to the inhibition of the EWS–FLI1-downstream Werner syndrome protein, which in turn made ES cell lines hypersensitive to the chemotherapeutic SN38, the active metabolite of irinotecan.Citation195 Utilizing this combination of trabectedin and subsequent SN38 treatment, Grohar et al were able to cause regression of two ES xeno-grafts with low drug concentrations and minimal toxicity.Citation195 A clinical trial with this combination has indeed begun, but results have not yet been posted.Citation196
IGFR1 and combination treatments
Insulin-dependent signal transduction plays an important role in the malignancy of ES. Prieur et al demonstrated that the fusion protein EWS–FLI1 can directly bind to the promoter and represses the expression of a regulator of the IGF-signaling pathway, IGFBP3, which is a tumor suppressor that disrupts interaction between the receptor IGFR1 and its ligand IGF, is crucial to prosurvival pathways.Citation197 Several anti-IGF1R-inhibiting antibodies have been investigated in clinical trials, and in addition to showing a 10% response rate in ES patients, they were generally well tolerated.Citation198,Citation199 Resistance to IGF1R inhibition, however, can occur,Citation200 and according to Lamhamedi-Cherradi et al, cells resistant to dalotuzumab, an IGFR1-inhibiting antibody, upregulated some mTOR pathway components. The combination of IGF1R and mTOR inhibition can synergistically bypass the resistance developed from single treatment of either agent and induce an antitumor response.Citation200 A clinical trial investigating this combination of IGFR1 and mTOR inhibition showed that 29% of ES patients had tumor reduction and two patients’ tumors regressed.Citation201 One patient, who had been previously treated with another IGF1R antibody but developed resistance, had a complete response with this combination treatment.Citation201
PARP inhibition
Another potential therapeutic target for treating ES is PARP, a chromatin-associated enzyme involved in DNA repair. Due to upregulation of PARP, ES has proved to be very sensitive to the PARP inhibitor olaparib.Citation202 Heske et al recently showed that combined inhibition of PARP and NAMPT, an enzyme crucial for the production of NAD+, a PARP substrate, resulted in delayed tumor growth and increased survival.Citation203 A Phase II clinical trial for PARP inhibition in refractory ES showed the treatment was safe and well tolerated, though there was no significant response, potentially because the small cohort consisted of patents who had previously been treated with chemotherapy.Citation204 Several ongoing clinical trials are examining the efficacy of PARP inhibition specifically in patients with defective DNA damage repair pathwaysCitation205 or in combination with other chemotherapeutic drugs.Citation206
ES: immunotherapy
Breakthroughs in immunobased treatments, notably with regard to PDL1/PD1-inhibiting antibodies, have led to successful treatments in several cancers. Recently, the FDA approved a fourth PDL1-specific antibody – durvalumab. Following are some of the major advances in immunotherapy for the treatment of ES.
Immunoblockage: PDL1 status and efficacy
Recently, Spurny et al discovered a lack of PDL1 expression in ES primary samples as well as established cell lines, though surprisingly over half the samples showed positive staining for the receptor – PD1.Citation192 Raj et al showed that 30% of tumors from ES patients expressed PDL1, and PDL1 expression was associated with treatment response.Citation207 Le et al recently showed that responsiveness to PDL1 inhibition was due to the lack of a mismatch repair mechanism regardless of tumor type,Citation208 raising the possibility of the mismatch repair pathway being implicated in the efficacy of this treatment in ES. A Phase II clinical trial has been set to investigate the efficacy of PDL1 inhibition in a variety of cancers, including ES with mismatch repair status as the selection criteria.Citation209
CHM1 and EZH2
CHM1, an endochondral bone protein, was reported by von Heyking et al to be highly expressed in ESCitation210,Citation211 and is implicated in stemness, enhanced proliferation, invasiveness, and metastasis.Citation210 CHM1 recognition by T cells can launch an immunoresponse against CHM1-expressing ES cells and significantly inhibit lung and liver metastatic burdens in vivo, suggesting the clinical potential of CHM1 as a target in ES treatment.Citation212 Another potential target is EZH2, a histone methyltransferase, shown by Thiel et al to induce an immunoresponse and ES-specific cytotoxicity when primed with allorestricted T cells.Citation211
Vaccine-based immunotherapy
A pilot study investigating the effect of the Vigil/FANG vaccine showed promising results.Citation213 Vigil/FANG consists of autologous tumor cells transfected with recombinant GM-CSF and short-hairpin RNA against furin.Citation213 When administered to the tumor site, GM-CSF can induce immunoresponses, whereas furin silencing can block activation of immune tolerance activity of TGFβ1/TGFβ2, ultimately resulting in tumor destruction.Citation213 Most of the participants had a complete knockdown of TGFβ1/TGFβ2, all had systemic tumor-specific immunoresponses, and it was estimated that 75% would survive past 1 year.Citation213 The vaccine was well tolerated, and this led to a Phase II clinical trial of the vaccine combined with two chemotherapeutic drugs: temozolomide and irinotecan.Citation214
Another ongoing clinical trial is looking into the efficacy of a vaccine containing CD25-depleted lymphocytes with tumor lysates and primed dendritic cells with or without IL7 in patients with high-risk ES.Citation215 Initial findings showed 57% of those vaccinated with immunostimulatory IL7 had a positive immunoresponse, and 40% of the patients were reported to be stable or without progressive disease.Citation215
Synovial sarcoma
SS gets its name from its microscopic similarity and proximity to the synovium, which is a specialized connective tissue that lines synovial joints, but in reality the development of tumor cells is not necessarily of synovial origin. While it is an STS typically found in the arms or legs and usually close to tendon sheaths and joint capsules, it can also occur in other locations, such as the heart, brain, and prostate. SS accounts for ~5%–10% of all STSCitation216 and 10%–20% of STS in adolescents and young adults;Citation217,Citation218 the median age of diagnosis is 35 years, though ages can range 5–85 years.Citation217,Citation219 Current standard of treatment includes surgery and radiotherapy, with SS displaying some sensitivity to chemotherapeutic agents like anthracyclines and ifosfamide.Citation217 Overall outcomes are poorer in adults, with the 5-year cancer-specific survival rate being 83% for children and adolescents, but only 62% in adults; even among adolescents and children, younger patients have better outcomes.Citation220 The extremities are the most common site of tumor origin, accounting for ~70% of cases;Citation220 ~50% of patients exhibit metastatic disease, with 74%–81% of those experiencing metastasis to the lungs.Citation216,Citation220 Smaller tumors and those restricted to the extremities portend better outcomes.Citation220 Despite differences in survival outcomes among varying ages, histological features of SS between children and adults seem to be identical.Citation220
Synovial sarcoma: subtypes
Although its cellular origin is unclear (its name is counterintuitive, because SS actually may not be of synovial origin),Citation216,Citation221 SS is generally divided into three histological subtypes: monophasic, biphasic, and poorly differentiated.Citation216,Citation220,Citation221 Monophasic SS is characterized by the presence of spindle cells and the absence or near-absence of glandular epithelial cells, whereas biphasic SS has equal presence of both spindle cells and glandular epithelial cells.Citation216,Citation221 In addition, monophasic SS displays fibrous and sarcomatous cells that are relatively uniform and small and form sheets. In contrast, biphasic SS presents with an epithelial appearance. Furthermore, poorly differentiated SS shows similarities to the small round cells found in ES.Citation216,Citation221 Another characteristic of SS is the unique chromosomal translocation (t[X;18]), which results in fusion of the SYT gene to the SSX1, SSX2, or, on rare occasions, the SSX4 gene.Citation217,Citation220,Citation222 A cytogenetic approach that makes use of reverse-transcription polymerase chain reactions can help to differentiate the monophasic and biphasic forms.
Synovial sarcoma: genomic landscape
Initially, SS was categorized in the miscellaneous soft-tissue tumor group by the World Health Organization, due to its unknown origin, despite the misleading name, as it bears no resemblance to synovial cells.Citation223,Citation224 The discovery of the translocation fusion SYT–SSX is a now a key characteristic of SS, with SYT–SSX1 found primarily in biphasic SS, but in rare cases also in monophasic SS, whereas SYT–SSX2 is found only in the monophasic subtype.Citation223,Citation225–Citation228 Those patients with the biphasic subtype appear to have longer survival than those with the monophasic counterpart, but more data are needed to corroborate these findings.Citation229 Similarly, those with SYT–SSX1 have better survival outcomes compared to those with the SYT–SSX2 fusion product.Citation229 Overall, 55% of all SS cases showed changes in the DNA CN and chromosome arms.Citation230 There is a higher frequency of genetic aberrations in monophasic subtypes (78% of cases) compared to only 16% and 5% of biphasic and poorly differentiated SS, respectively.Citation230 ERBB2 and IGFBP2 are genes highly expressed in the epithelial region of the biphasic subtype, potentially indicating the involvement of these genes in epithelial differentiation programs in the biphasic subtypes.Citation231 Additionally, E-cadherin and α-catenin are preserved in the epithelial components of the biphasic subtype and are correlated with longer survival rates.Citation232 Expression of E-cadherin is also associated with low mitotic rates.Citation232
The most frequent gains occur on chromosomes 2, 8, and 12,Citation230 and in particular comprise MDM2, MSH2, KCNK12, DCC, CDK2, ERBB3, SAS, and CDK4.Citation229 Conversely, the most frequent losses occur on chromosomes 3 and 13,Citation230 with common losses of HRAS, RASSF1, and CCND1.Citation229 Over 50% of SS cases show positive expression for EGFR, over 40% express SALL2, and the majority show the presence of Bcl2 (91%), pancytokeratin (77%), EMA (75%), and cytokeratin 7.Citation233 Using a cDNA microarray, Nagayama et al discovered that SS clustered together with another STS type: malignant peripheral nerve sheath tumors (MPNSTs).Citation223 However, Terry et al later showed that TLE staining can differentiate SS from MPNST: >90% of SS samples expressed TLE, but <5% of MPNSTs stained positively for TLE.Citation234 Nagayama et al also discovered some commonly upregulated genes in SS to be crucial for neural crest development, migration, and differentiation, suggesting the potential origin of SS was derived from neural crest cells.Citation223
Synovial sarcoma: targeted therapeutics
Several targeted therapies are currently being tested in clinical trials.Citation235–Citation238 One trial that is currently recruiting patients is examining the efficacy of inhibiting glucose-6-phosphate dehydrogenase, which is crucial for tumorigenesis of SS.Citation235 Two clinical trials, one for pediatric patients and the other for adults, are investigating the effectiveness of inhibiting a polycomb group protein – EZH2.Citation236,Citation237 Another trial is investigating the effect of inhibiting the angiogenic receptors VEGFR2 and VEGFR3.Citation238 Several clinical trials are also examining combination regimens.Citation239,Citation240 One investigated the combination of an mTOR inhibitor, everolimus, and a multiple TKR inhibitor (c-Kit and PDGFR) – imatinib. The findings were quite promising: of nine participants analyzed, five showed stable disease and four had progression, for a response rate of 55%.Citation239 A trial of dacarbazine, a chemotherapeutic agent, together with sorafenib, an Raf/VEGFR2/PDGFRβ inhibitor, resulted in a response rate of >70%: 5 patients had partial responses, 22 had stable disease, and 8 had disease progression, with one death due to adverse effects.Citation240
Synovial sarcoma: immunotherapy
The use of the SYT–SSX fusion product as a target for immunotherapy in SS has shown some therapeutic benefit.Citation241–Citation243 Suminoe et al showed that vaccination of dendritic cells armed with the SYT–SSX peptide was well tolerated, with no adverse effects in one patient whose disease had been stable for 2 months.Citation242 Similarly, though using only the fusion peptide, Kawaguchi et al also demonstrated a patient’s tolerance to the vaccine. The patient had no disease progression, along with a decrease in circulating cytotoxic T lymphocytes, suggesting these were potentially localized at the site of the tumor, though no biopsy was performed.Citation241 Kawaguchi et al further demonstrated the efficacy of the fusion peptide vaccine in conjunction with the administration of an immunostimulant – IFNα.Citation243 Half the patient cohort had stable disease, as opposed to only 11% of patients in the vaccine-only arm.Citation243 One caveat of these clinical trials is small samples, owing to the rarity of this type of sarcoma, making it difficult to see the full potential in utilizing the fusion peptide as a therapeutic target.
NY-ESO1 is a tumor antigen belonging to the testis family that is highly expressed in malignant tissues and in the testis, but not in other normal tissues.Citation244,Citation245 Over half of SS patients express NY-ESO1,Citation244 and because it is immunogenic,Citation246–Citation248 it is an attractive target for immunotherapy. In a first-ever clinical trial looking into the effect of administering NY-ESO1 receptor-expressing autologous T cells in patients with metastatic SS, Robbins et al reported a response rate of >60%.Citation245 Four of six patients showed partial responses, with the longest being 18 months, and several individuals also had regressed lung metastases.Citation245 The treatment was well tolerated, leading to the initiation of other similar clinical trials looking into the effectiveness of NY-ESO1 in immunotherapy.Citation249–Citation251 One currently active clinical trial is examining the efficacy of combining atezolizumab, a PDL1 inhibitor, with a dendritic cell-specific vector containing the gene for NY-ESO1.Citation252 Another clinical trial took a different approach in that allogeneic tumor lysate expression using NY-ESO1 was used to prime patients’ autologous dendritic cells.Citation253 Upon completion, and depending upon the results, these investigational new therapies could be used in the clinic as treatment measures for SS patients.
Epigenetic changes in STS clinical samples
Epigenetic regulators are more recently identified therapeutic targets for STS, and small-molecule inhibitors that inhibit them could prove to be useful in treating the disease. In particular, such regulators as DNA methyltransferases, histone deacetylases (HDACs), and the histone-modifying enzyme EZH2 have been implicated in the pathogenesis of several types of STS.Citation254 Kawaguchi et alCitation255 used clinical samples to demonstrate that the promoter methylation status of tumor-related genes could have an association with the pathogenesis of STS. Currently, however, no DNA methylation inhibitors have been tested for use in STS.
Histone modifications like phosphorylation, acetylation, methylation, and ubiquitination are crucial in the regulation of genes, including oncogenes and tumor suppressors. In particular, acetylation changes modulated by HDAC occur in several malignancies, including STS. Preclinical treatment with PCI24781, an HDAC inhibitor, has been shown to inhibit growth and cause apoptosis in several STS cell lines, including SS and uterine sarcoma.Citation254 Also, EZH2 is an important histone methyltransferase and, when overexpressed in STS samples, is correlated with greater tumor size, metastases, and poor progression.Citation256 In particular, EZH2 mediates the expression of the tumor suppressor ERG1 via SYT-SSX in SS.Citation256 Also, elevated levels of EZH2 are found in RMS and ES.Citation257 As such, EZH2 inhibition might prove successful as a therapeutic approach for STS. Currently, the EZH2 inhibitor tazemetostat is being tested in a Phase II multicenter clinical trial for patients with SS.Citation257
Conclusion
Given the genetic and histological diversity of this large family of cancers, the management of adult STS calls for a multidisciplinary approach to achieve optimal outcomes. Over the past 30 years, our knowledge of STS biology has progressed very little when compared with other malignancies. This trend has slowly started to change, and additional developments could lead to new therapeutic strategies and treatment options. Several clinical investigations of therapies outlined in this review for STS are under way, and these summarized in .
Disclosure
The authors report no conflicts of interest in this work.
References
- JemalATiwariRCMurrayTCancer statistics, 2004CA Cancer J Clin20045482914974761
- SiegelRLMillerKDJemalACancer statisticsCA Cancer J Clin20156552925559415
- LawrenceWJrDoneganWLNtarajanNMettlinCBeartRWinchesterDAdult soft tissue sarcomas: a pattern of care survey of the American College of SurgeonsAnn Surg19872053493593566372
- Christie-LargeMJamesSLTiessenLDaviesAMGrimerRJImaging strategy for detecting lung metastases at presentation in patients with soft tissue sarcomasEur J Cancer2008441841184518640829
- American Cancer SocietySurvival by stage of soft tissue sarcoma2017 Available from: https://www.cancer.org/cancer/soft-tissue-sarcoma/detection-diagnosis-staging/survival-rates.htmlAccessed March 28, 2018
- EdgeSBByrdDRComptonCCFritzAGGreeneFLTrottiACancer Staging Manual7th edHeidelbergSpringer2010
- ZagarsGKBalloMTPistersPWPrognostic factors for patients with localized soft-tissue sarcoma treated with conservation surgery and radiation therapy: an analysis of 1225 patientsCancer2003972530254312733153
- FletcherCDBridgeJAHogendoornPCMertensFWHO Classification of Tumours of Soft Tissue and Bone4th edGenevaWorld Health Organization2013
- KurodaMIshidaTHoriuchiHChimeric TLS/FUS-CHOP gene expression and the heterogeneity of its junction in human myxoid and round cell liposarcomaAm J Pathol1995147122112277485386
- CragoASocciNDDeCarolisPCopy number losses define subgroups of dedifferentiated liposarcoma with poor prognosis and genomic instabilityClin Cancer Res20021813341340
- BellTWhat is liposarcoma?2012 Available from: http://sarcomahelp.org/liposarcoma.htmlAccessed March 28, 2018
- CragoAMSingerSClinical and molecular approaches to well-differentiated and dedifferentiated liposarcomaCurr Opin Oncol20112337337821552124
- KanojiaDNagataYGargMGenomic landscape of liposarcomaOncotarget20156424294244426643872
- RiekerRJWeitzLehnerBGenomic profiling reveals subsets of dedifferentiated liposarcoma to follow separate molecular pathwaysVirchows Arch201045627728520039060
- AntonescuCRTschernyavskySJDecusearaRPrognostic impact of P53 status, TLS-CHOP fusion transcript structure, and histological grade in myxoid liposarcoma: a molecular and clinicopathologic study of 82 casesClin Cancer Res200173977398711751490
- RiekerRJJoosSBartschCDistinct chromosomal imbalances in pleomorphic and in high-grade dedifferentiated liposarcomasInt J Cancer200299687311948494
- Spinella-JaegleSRawadiGKawaiSSonic hedgehog increases the commitment of pluripotent mesenchymal cells into osteoblastic lineage and abolishes adipocytic differentiationJ Cell Sci20011142085209411493644
- GobbleRMQinLXBrillERExpression profiling of liposarcoma yields a multigene predictor of patient outcome and identifies genes that contribute to liposarcomagenesisCancer Res2011712697270521335544
- CostaADaidoneMGDapraiLTelomere maintenance mechanisms in liposarcomas: association with histologic subtypes and disease progressionCancer Res2006668918892416951210
- ItalianoABianchiniLGjernesEClinical and biological significance of CDK4 amplification in well-differentiated and dedifferentiated liposarcomasClin Cancer Res2009155696570319737942
- TapWDEilberFCGintherCEvaluation of well-differentiated/de-differentiated liposarcomas by high-resolution oligonucleotide array-based comparative genomic hybridizationGenes Chromosomes Cancer2011509511221117066
- TaylorBSDeCarolisPLAngelesCVFrequent alterations and epigenetic silencing of differentiation pathway genes in structurally rearranged liposarcomasCancer Discov2011158759722328974
- SingerSSocciNDAmbrosiniGGene expression profiling of liposarcoma identifies distinct biological types/subtypes and potential therapeutic targets in well-differentiated and dedifferentiated liposar-comaCancer Res2007676626663617638873
- CoindreJMPédeutourFAuriasAWell-differentiated and dedifferentiated liposarcomasVirchows Arch201045616717919688222
- SchmidtHBartelFKapplerMGains of 13q are correlated with a poor prognosis in liposarcomaMod Pathol20051863864415540119
- KoelscheCRennerMHartmannWTERT promoter hotspot mutations are recurrent in myxoid liposarcomas but rare in other soft tissue sarcoma entitiesJ Exp Clin Cancer Res2014333324726063
- SreekantaiahCThe cytogenetic and molecular characterization of benign and malignant soft tissue tumorsCytogenet Cell Genet19988213299763652
- FritzBSchubertFWrobelGMicroarray-based copy number and expression profiling in dedifferentiated and pleomorphic liposarcomaCancer Res2002622993299812036902
- SchoffskiPRay-CoquardILCioffiAActivity of eribulin mesylate in patients with soft-tissue sarcoma: a phase 2 study in four independent histological subtypesLancet2011121045105221937277
- SchoffskiPChawlaSMakiRGEribulin versus dacarbazine in previously treated patients with advanced liposarcoma or leiomyosarcoma: a randomised, open-label, multicenter, phase 3 trialLancet20163871629163726874885
- GrossoFJonesRLDemetriGDEfficacy of trabectedin in advanced pretreated myxoid liposarcomas: a retrospective studyLancet Oncol2007859560217586092
- GiandomenicoSDFrapolliRBelloEMode of action of trabectedin in myxoid liposarcomaOncogene2014335201521024213580
- DicksonMATapWDKeohanMLPhase II trial of the CDK4 inhibitor PD0332991 in patients with advanced CDK4-amplified well-differentiated or dedifferentiated liposarcomaJ Clin Oncol2013312024202823569312
- Memorial Sloan Kettering Cancer CenterStudy of abemaciclib in dedifferentiated liposarcoma Available from: https://clinicaltrials.gov/ct2/show/NCT02846987. NLM identifier: NCT02846987Accessed March 28, 2018
- Hadassah Medical OrganizationA phase II single arm study assessing efficacy and safety of ribociclib in patients with advanced well-differentiated or dedifferentiated liposarcoma Available from: https://clinicaltrials.gov/ct2/show/NCT02571829. NLM identifier: NCT02571829Accessed March 28, 2018
- Vector OncologyA phase II study of pazopanib in the treatment of surgically unresectable or metastatic liposarcoma Available from: https://clinicaltrials.gov/ct2/show/NCT01506596. NLM identifier: NCT01506596Accessed March 28, 2018
- Grupo Espanol de Investigacion en SarcomasPhase II clinical trial of pazopanib to evaluate the activity and tolerability in patients with advanced and/or metastatic liposarcoma who have relapsed following standard therapies or for whom no standard therapy exists Available from: https://clinicaltrials.gov/ct2/show/NCT01692496. NLM identifier: NCT01692496Accessed March 28, 2018
- Northwestern UniversityA phase II study of pazopanib with oral topotecan in patients with metastatic and non-resectable soft tissue and bone sarcomas Available from: https://clinicaltrials.gov/ct2/show/NCT02357810. NLM identifier: NCT02357810Accessed March 28, 2018
- ChawlaSPStaddonAPBakerLHPhase II study of the mammalian target of rapamycin inhibitor ridaforolimus in patients with advanced bone and soft tissue sarcomasJ Clin Oncol201230788422067397
- MD Anderson Cancer CenterStudy of neoadjuvant checkpoint blockade in patients with surgically resectable undifferentiated pleomorphic sarcoma and dedifferentiated liposarcoma Available from: https://clinicaltrials.gov/ct2/show/NCT03307616. NLM identifier: NCT03307616Accessed March 28, 2018
- National Cancer InstituteRandomized phase II study of nivolumab with or without ipilimumab in patients with metastatic or unresectable sarcoma Available from: https://clinicaltrials.gov/ct2/show/NCT02500797. NLM identifier: NCT02500797Accessed March 28, 2018
- Fox Chase Cancer CenterPhase II trial of ribociclib and everolimus in advanced dedifferentiated liposarcoma (DDL) and leiomyosarcoma (LMS) Available from: https://clinicaltrials.gov/ct2/show/NCT03114527. NLM identifier: NCT03114527Accessed March 28, 2018
- AdaptimmuneA pilot study of NY-ESO-1c259 T cells in subjects with advanced myxoid/round cell liposarcoma Available from: https://clinicaltrials.gov/ct2/show/NCT02992743. NLM identifier: NCT02992743Accessed March 28, 2018
- Fred Hutchinson Cancer Research CenterAvelumab and trabectedin in treating patients with liposarcoma or leiomyosarcoma that is metastatic or cannot be removed Available from: https://clinicaltrials.gov/ct2/show/NCT03074318. NLM identifier: NCT03074318Accessed March 28, 2018
- Immune DesignA randomized, open-label, phase 2 trial of CMB305 (sequentially administered LV305 and G305) and atezolizumab in patients with locally advanced, relapsed, or metastatic sarcoma expressing NY-ESO-1 Available from: https://clinicaltrials.gov/ct2/show/NCT02609984. NLM identifier: NCT02609984Accessed March 28, 2018
- Sarcoma Oncology Research CenterA phase 1B single center investigation of safety/efficacy of nivolumab (Opdivo) and ABI-009 (nab-rapamycin) in patients with advanced undifferentiated pleomorphic sarcoma, liposarcoma, chondrosarcoma, osteosarcoma and Ewing sarcoma Available from: https://clinicaltrials.gov/ct2/show/NCT03190174. NLM identifier: NCT03190174Accessed March 28, 2018
- Washington University School of MedicineA non-inferiority study of doxorubicin with upfront dexrazoxane plus olaratumab for the treatment of advanced or metastatic soft tissue sarcoma Available from: https://clinicaltrials.gov/ct2/show/NCT02584309. NLM identifier: NCT02584309Accessed March 28, 2018
- LammWNatterCSchurSDistinctive outcome in patients with non-uterine and uterine leiomyosarcomaBMC Cancer20141498125523155
- YoungRJBrownNJReedMWHughesDWollPJAngiosarcomaLancet Oncol20101198399120537949
- FayetteJMartinEPiperno-NeumannSAngiosarcomas, a heterogeneous group of sarcomas with specific behavior depending on primary site: a retrospective study of 161 casesAnn Oncol2007182030203617974557
- StrobbeLJPeterseHLvan TinterenHWijnmaalenARutgersEJAngiosarcoma of the breast after conservation therapy for invasive cancer, the incidence and outcome: an unforseen [sic] sequelaBreast Cancer Res Treat1998471011099497098
- MuraliRChandramohanRMöllerITargeted massively parallel sequencing of angiosarcomas reveals frequent activation of the mitogen activated protein kinase pathwayOncotarget20156360413605226440310
- ItakuraEYamamotoHOdaYTsuneyoshiMDetection and characterization of vascular endothelial growth factors and their receptors in a series of angiosarcomasJ Surg Oncol200897748118041747
- ItalianoAChenCLThomasRAlterations of the p53 and PIK3CA/AKT/mTOR pathways in angiosarcomas: a pattern distinct from other sarcomas with complex genomicsCancer20121185878588722648906
- MannerJRadlwimmerBHohenbergerPMYC high level gene amplification is a distinctive feature of angiosarcomas after irradiation or chronic lymphedemaAm J Pathol2010176343920008140
- ShonWSukovWRJenkinsSMFolpeALMYC amplification and overexpression in primary cutaneous angiosarcoma: a fluorescence in-situ hybridization and immunohistochemical studyMod Pathol20142750951524091875
- LaéMLebelAHamel-ViardFCan c-Myc amplification reliably discriminate postradiation from primary angiosarcoma of the breast?Cancer Radiother20151916817425863565
- FernandezAPSunYTubbsRRGoldblumJRBillingsSDFISH for MYC amplification and anti-MYC immunohistochemistry: useful diagnostic tools in the assessment of secondary angiosarcoma and atypical vascular proliferationsJ Cutan Pathol20123923424222121953
- GinterPSMosqueraJMMacDonaldTYD’AlfonsoTMRubinMAShinSJDiagnostic utility of MYC amplification and anti-MYC immunohistochemistry in atypical vascular lesions, primary or radiation-induced mammary angiosarcomas, and primary angiosarcomas of other sitesHum Pathol201445470971624457083
- GuoTZhangLChangNESingerSMakiRGAntonescuCRConsistent MYC and FLT4 gene amplification in radiation-induced angiosarcoma but not in other radiation-associated atypical vascular lesionsGenes Chromosomes Cancer201150253320949568
- MentzelTSchildhausHUPalmedoGBüttnerRKutznerHPostradiation cutaneous angiosarcoma after treatment of breast carcinoma is characterized by MYC amplification in contrast to atypical vascular lesions after radiotherapy and control cases: clinicopathological, immunohistochemical and molecular analysis of 66 casesMod Pathol201225758521909081
- TolaniBGopalakrishnanRPunjVTargeting Myc in KSHV-associated primary effusion lymphoma with BET bromodomain inhibitorsOncogene201429332928293723792448
- PottiAGantiAKTendulkarKDetermination of vascular endothelial growth factor (VEGF) overexpression in soft tissue sarcomas and the role of overexpression in leiomyosarcomaJ Cancer Res Clin Oncol2004130525614600832
- PottiAGantiAKFosterHImmunohistochemical detection of HER-2/neu, c-Kit (CD117) and vascular endothelial growth factor (VEGF) overexpression in soft tissue sarcomasAnticancer Res20042433333715015617
- PakosEEGoussiaACTsekerisPGPapachristouDJStefanouDAgnantisNJExpression of vascular endothelial growth factor and its receptor, KDR/Flk-1, in soft tissue sarcomasAnticancer Res2005253591359616101185
- ChaoCAl-SaleemTBrooksJJRogatkoAKraybillWGEisenbergBVascular endothelial growth factor and soft tissue sarcomas: tumor expression correlates with gradeAnn Surg Oncol2001826026711314944
- RanieriGMammìMDi PaolaEEPazopanib a tyrosine kinase inhibitor with strong anti-angiogenetic activity: a new treatment for metastatic soft tissue sarcomaCrit Rev Oncol Hematol20148932232924041629
- AntonescuCRYoshidaAGuoTKDR activating mutations in human angiosarcomas are sensitive to specific kinase inhibitorsCancer Res2009697175717919723655
- AmoYMasuzawaMHamadaYKatsuokaKExpression of vascular endothelial growth factor in a human hemangiosarcoma cell line (ISO-HAS)Arch Dermatol Res200129329630111480589
- BehjatiSTarpeyPSSheldonHRecurrent PTPRB and PLCG1 mutations in angiosarcomaNat Genet20144637637924633157
- MellbergSDimbergABahramFTranscriptional profiling reveals a critical role for tyrosine phosphatase VE-PTP in regulation of VEGFR2 activity and endothelial cell morphogenesisFASEB J2009231490150219136612
- RaviVPatelSVascular sarcomasCurr Oncol Rep20131534735523852636
- von MehrenMRankinCGoldblumJRPhase 2 Southwest Oncology Group-directed intergroup trial (S0505) of sorafenib in advanced soft tissue sarcomasCancer201211877077621751200
- PenelNRay-CoquardIBal-MahieuCLow level of baseline circulating VEGF-A is associated with better outcome in patients with vascular sarcomas receiving sorafenib: an ancillary study from a phase II trialTarget Oncol2014927327724218035
- HurwitzHIDowlatiASainiSPhase I trial of pazopanib in patients with advanced cancerClin Cancer Res2009154220422719509175
- van der GraafWTBlayJYChawlaSPPazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trialLancet201227918791886
- VerweijJSleijferSPazopanib, a new therapy for metastatic soft tissue sarcomaExpert Opin Pharmacother20131492993523488774
- OgataDYanagisawaHSuzukiKOashiKYamazakiNTsuchidaTPazopanib treatment slows progression and stabilizes disease in patients with taxane-resistant cutaneous angiosarcomaMed Oncol20163311627613162
- RosenAThimonSTernantDMachetMCPaintaudGMachetLPartial response to bevacizumab of an extensive cutaneous angiosarcoma of the faceBr J Dermatol201016322522720394623
- AgulnikMYarberJLOkunoSHAn open-label, multicenter, phase II study of bevacizumab for the treatment of angiosarcoma and epithelioid hemangioendotheliomasAnn Oncol20132425726322910841
- KoontzBFMilesEFRubioMAPreoperative radiotherapy and bevacizumab for angiosarcoma of the head and neck: two case studiesHead Neck200830262617685450
- FullerCKCharlsonJADankleSKRussellTJDramatic improvement of inoperable angiosarcoma with combination paclitaxel and bevacizumab chemotherapyJ Am Acad Dermatol201063e83e8420846560
- DicksonMAD’AdamoDRKeohanMLPhase II trial of gemcitabine and docetaxel with bevacizumab in soft tissue sarcomaSarcoma2015201553247826074722
- MakiRGJungbluthAAGnjaticSA pilot study of anti-CTLA4 antibody ipilimumab in patients with synovial sarcomaSarcoma2013201316814523554566
- SindhuSGimberLHCranmerLMcBrideAKraftASAngiosarcoma treated successfully with anti-PD-1 therapy: a case reportJ Immunother Cancer201755828716069
- LammWNatterCSchurSDistinctive outcome in patients with non-uterine and uterine leiomyosarcomaBMC Cancer20141498125523155
- GuoXJoVYMillsAMClinically relevant molecular subtypes in leiomyosarcomaClin Cancer Res2015213501251125896974
- HernandoECharytonowiczEDudasMEThe AKT-mTOR pathway plays a critical role in the development of leiomyosarcomaNat Med20071374875317496901
- ShiGPerleMAMittalKLet-7 repression leads to HMGA2 overexpression in uterine leiomyosarcomaJ Cell Mol Med2009133898390519602040
- HayashiTHoriuchiASanoKPotential Role of LMP2 as tumor-suppressor defines new targets for uterine leiomyosarcoma therapySci Rep2011118022355695
- ShanWAkinfenwaPYSavannahKBA small-molecule inhibitor targeting the mitotic spindle checkpoint impairs the growth of uterine leiomyosarcomaClin Cancer Res2012183352336522535157
- SetiaAKanotraSAggarwalRBhavthankarDPEpithelioid leiomyosarcoma of uterusBMJ Case Rep2012
- EdrisBWeiskopfKVolkmerAKAntibody therapy targeting the CD47 protein is effective in a model of aggressive metastatic leiomyosarcomaProc Natl Acad Sci U S A20121096656666122451919
- HuJRaoUNJasaniSKhannaVYawKSurtiULoss of DNA copy number of 10q is associated with aggressive behavior of leiomyosarcoma: a comparative genomic hybridization studyCancer Genet Cytogenet2005161202716080954
- HuJKhannaVJonesMSurtiUGenomic alterations in uterine leiomyosarcomas: potential markers for clinical diagnosis and prognosisGenes Chromosomes Cancer20013111712411319799
- PonceletCWalkerFMedelenatPExpression of CD44 standard and isoforms V3 and V6 in uterine smooth muscle tumors: a possible diagnostic tool for the diagnosis of leiomyosarcomaHum Pathol20093211901196
- SavannahKJDemiccoEGDual targeting of mTOR and aurora-A kinase for the treatment of uterine leiomyosarcomaClin Cancer Res2012184633464522821997
- SchvartzmanJMSotilloRBenezraBMitotic chromosomal instability and cancer: mouse modeling of the human diseaseNat Rev Cancer20101010211520094045
- EdrisBEspinosaIMühlenburgTROR2 is a novel prognostic biomarker and a potential therapeutic target in leiomyosarcoma and gastrointestinal stromal tumourJ Pathol201222722323322294416
- GiraudetALBadelJNCassierPSYNFRIZZ: a phase IA/IB of a radiolabelled monoclonal AB for the treatment of relapsing synovial sarcomaJ Nucl Med20145522324434294
- SetteGSalvatiVMemeoLEGFR inhibition abrogates leiomyosarcoma cell chemoresistance through inactivation of survival pathways and impairment of CSC potentialPLoS One2012710e4689123056514
- HayashiTFaustmanDLDevelopment of spontaneous uterine tumors in low molecular mass polypeptide-2 knockout miceCancer Res200262242711782352
- MakiRGKraftASScheuKA multicenter phase II study of bortezomib in recurrent or metastatic sarcomasCancer20051031431143815739208
- GaoCFXieQZhangYWTherapeutic potential of hepatocyte growth factor/scatter factor neutralizing antibodies: inhibition of tumor growth in both autocrine and paracrine hepatocyte growth factor/scatter factor – c-Met-driven models of leiomyosarcomaMol Cancer Ther200982803281019825800
- BurgessTCoxonAMeyerSFully human monoclonal antibodies to hepatocyte growth factor with therapeutic potential against hepatocyte growth factor/c-Met-dependent human tumorsCancer Res2006661721172916452232
- IvesonTDonehowerRCDavidenkoIRilotumumab in combination with epirubicin, cisplatin, and capecitabine as first-line treatment for gastric or oesophagogastric junction adenocarcinoma: an open-label, dose de-escalation phase B study and a double-blind, randomized phase 2 studyLancet Oncol2014151007101824965569
- AmgenA multicenter, phase 1/1B, open label study evaluating the safety, tolerability and pharmacokinetics of rilotumumab in Japanese subjects Available from: https://clinicaltrials.gov/ct2/show/NCT01791374. NLM identifier: NCT01791374Accessed March 28, 2018
- Gynecologic Oncology GroupA phase II evaluation of AMG 102 (rilotumumab) (NSC #750009) in the treatment of persistent or recurrent epithelial ovarian, fallopian tube, or primary peritoneal carcinoma Available from: https://clinicaltrials.gov/ct2/show/NCT01039207. NLM identifier: NCT01039207Accessed March 28, 2018
- University of PittsburghA phase I/II trial of AMG 102 and erlotinib in previously treated patients with advanced non-small cell lung cancer Available from: https://clinicaltrials.gov/ct2/show/NCT01233687. NLM identifier: NCT01233687Accessed March 28, 2018
- Duke University Medical CenterPhase II study to evaluate the efficacy and safety of AMG 102 and Avastin in subjects with recurrent malignant glioma Available from: https://clinicaltrials.gov/ct2/show/NCT01113398. NLM identifier: NCT01113398Accessed March 28, 2018
- Southwest Oncology GroupA biomarker-driven master protocol for previously treated squamous cell lung cancer (Lung-MAP) Available from: https://clinicaltrials.gov/ct2/show/NCT02154490. NLM identifier: NCT02154490Accessed March 28, 2018
- PaoluzziLCacavioAGhesaniMResponse to anti-PD1 therapy with nivolumab in metastatic sarcomasClin Sarcoma Res201662428042471
- Royal Marsden NHS Foundation TrustA phase I study to assess the safety and tolerability of pembrolizumab in combination with fixed rate gemcitabine chemotherapy in patients with leiomyosarcoma and undifferentiated pleomorphic sarcoma Available from: https://clinicaltrials.gov/ct2/show/NCT03123276. NLM identifier: NCT03123276Accessed March 28, 2018
- Institut BergoniéCombination of MK3475 and metronomic cyclophosphamide in patients with advanced sarcomas: multicentre phase II trial Available from: https://clinicaltrials.gov/ct2/show/NCT02406781. NLM identifier: NCT02406781Accessed March 28, 2018
- HeineAKristiansenGSchildHHBrossartPSuccessful treatment of refractory leiomyosarcoma with PD-1 inhibitor nivolumabAnn Oncol2016271813181427329248
- GeorgeSMiaoDDemetriGDLoss of PTEN is associated with resistance to anti-PD-1 checkpoint blockade therapy in metastatic uterine leiomyosarcomaImmunity20174619720428228279
- EdrisBWeiskopfKWeissmanILvan de RijnMFlipping the script on macrophages in leiomyosarcomaOncoimmunology201211202120423170280
- Forty SevenA phase I dose escalation trial of the humanized anti-CD47 monoclonal antibody Hu5F9-G4 in haematological malignancies (CAMELLIA) Available from: https://clinicaltrials.gov/ct2/show/NCT02678338. NLM identifier: NCT02678338Accessed March 28, 2018
- CelgeneA phase 1, open-label, dose finding study of CC-90002, a monoclonal antibody directed against CD47, in subjects with acute myeloid leukemia and high-risk myelodysplastic syndrome Available from: https://clinicaltrials.gov/ct2/show/NCT02641002. NLM identifier: NCT02641002Accessed March 28, 2018
- Forty SevenA first-in-human phase 1 dose escalation trial of Hu5F9-G4 in patients with advanced solid malignancies Available from: https://clinicaltrials.gov/ct2/show/NCT02216409. NLM identifier: NCT02216409Accessed March 28, 2018
- Trillium TherapeuticsA phase 1A/1B dose escalation and expansion trial of TTI-621, a novel biologic targeting CD47, in subjects with relapsed or refractory hematologic malignancies and selected solid tumors Available from: https://clinicaltrials.gov/ct2/show/NCT02663518. NLM identifier: NCT02663518Accessed March 28, 2018
- CelgeneA phase I, open-label, dose finding study of CC-90002, a monoclonal antibody directed against CD47, in subjects with advanced solid and hematologic cancers Available from: https://clinicaltrials.gov/ct2/show/NCT02367196. NLM identifier: NCT02367196Accessed March 28, 2018
- Alexo TherapeuticsA phase 1, dose escalation study of ALX148 in patients with advanced solid tumors and lymphoma Available from: https://clinicaltrials.gov/ct2/show/NCT03013218. NLM identifier: NCT03013218Accessed March 28, 2018
- XiaSJPresseyJGBarrFGMolecular pathogenesis of rhabdomyosarcomaCancer Biol Ther200219710412170781
- HuhWWSkapekSXChildhood rhabdomyosarcoma: new insight on biology and treatmentCurr Oncol Rep20101240241020820958
- OgnjanovicSLinaberyAMCharbonneauBRossJATrends in childhood rhabdomyosarcoma incidence and survival in the United States, 1975–2005Cancer20091154218422619536876
- HinikerSMDonaldsonSSRecent advances in understanding and managing rhabdomyosarcomaF1000Prime Rep201575926097732
- NewtonWAGehanEAWebberBLClassification of rhabdomyosarcomas and related sarcomas: pathologic aspects and proposal for a new classification – an Intergroup Rhabdomyosarcoma StudyCancer199576107310858625211
- TerrierPHenry-AmarMTricheTJIs neural-ectodermal differentiation of Ewing’s sarcoma of bone associated with an unfavorable prognosis?Eur J Cancer199531A3073147786593
- HosoiHCurrent status of treatment for pediatric rhabdomyosarcoma in the USA and JapanPediatr Int201658818726646016
- MalempatiSHawkinsDSRhabdomyosarcoma: review of the Children’s Oncology Group (COG) Soft-Tissue Sarcoma Committee experience and rationale for current COG studiesPediatr Blood Cancer20125951022378628
- ShernJFChenLChmieleckiJComprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumorsCancer Discov2014421623124436047
- HuhWWSkapekSXChildhood rhabdomyosarcoma: new insight on biology and treatmentCurr Oncol Rep20101240241020820958
- SorensenPHLynchJCQualmanSJPAX3-FKHR and PAX7-FKHR gene fusions are prognostic indicators in alveolar rhabdomyosarcoma: a report from the Children’s Oncology GroupJ Clin Oncol2002202672267912039929
- FredericksWJGaliliNMukhopadhyaySThe PAX3-FKHR fusion protein created by the t(2;13) translocation in alveolar rhabdomyosarcomas is a more potent transcriptional activator than PAX3Mol Cell Biol199515152215357862145
- AndersonMJSheltonGDCaveneeWKArdenKCEmbryonic expression of the tumor-associated PAX3-FKHR fusion protein interferes with the developmental functions of Pax3Proc Natl Acad Sci U S A2001981589159411171995
- EpsteinJASongBLakkisMWangCTumor-specific PAX3-FKHR transcription factor, but not PAX3, activates the platelet-derived growth factor alpha receptorMol Cell Biol199818411841309632796
- LagutinaIConwaySJSublettJGrosveldGFPax3-FKHR knock-in mice show developmental aberrations but do not develop tumorsMol Cell Biol2002227204721612242297
- CroseLEGalindoKAKephartJGAlveolar rhabdomyosarcomaassociated PAX3-FOXO1 promotes tumorigenesis via Hippo pathway suppressionJ Clin Invest201412428529624334454
- LinardicCMNainiSHerndonJE2ndKesserwanCQualmanSJCounterCMThe PAX3-FKHR fusion gene of rhabdomyosarcoma cooperates with loss of p16INK4A to promote bypass of cellular senescenceCancer Res2007676691669917638879
- WilliamsonDMissiagliaEde ReyniesAFusion gene-negative alveolar rhabdomyosarcoma is clinically and molecularly indistinguishable from embryonal rhabdomyosarcomaJ Clin Oncol2010282151215820351326
- BarrFGDuanFSmithLMGenomic and clinical analyses of 2p24 and 12q13–14 amplification in alveolar rhabdomyosarcoma: a report from the Children’s Oncology GroupGenes Chromosomes Cancer20094866167219422036
- LohWEJrScrableHJLivanosEHuman chromosome 11 contains two different growth suppressor genes for embryonal rhabdomyosarcomaProc Natl Acad Sci U S A199289175517591347425
- Weber-HallSAndersonJMcManusAGains, losses, and amplification of genomic material in rhabdomyosarcoma analyzed by comparative genomic hybridizationCancer Res199656322032248764111
- BridgeJALiuJWeiboltVNovel genomic imbalances in embryonal rhabdomyosarcoma revealed by comparative genomic hybridization and fluorescence in situ hybridization: an intergroup rhabdomyosarcoma studyGenes Chromosomes Cancer20002733734410719362
- GordonTMcManusAAndersonJCytogenetic abnormalities in 42 rhabdomyosarcoma: a United Kingdom Cancer Cytogenetics Group StudyMed Pediatr Oncol20013625926711452933
- StrattonMRFisherCGustersonBACooperCSDetection of point mutations in N-ras and K-ras genes of human embryonal rhabdomyosarcomas using oligonucleotide probes and the polymerase chain reactionCancer Res198949632463272680062
- PresseyJGAndersonJRCrossmanDKLynchJCBarrFGHedgehog pathway activity in pediatric embryonal rhabdomyosarcoma and undifferentiated sarcoma: a report from the Children’s Oncology GroupPediatr Blood Cancer20115793093821618411
- PaulsonVChandlerGRakhejaDHigh-resolution array CGH identifies common mechanisms that drive embryonal rhabdomyosarcoma pathogenesisGenes Chromosomes Cancer20115039740821412928
- ShuklaNAmeurNYilmazIOncogene mutation profiling of pediatric solid tumors reveals significant subsets of embryonal rhabdomyosarcoma and neuroblastoma with mutated genes in growth signaling pathwaysClin Cancer Res20121874875722142829
- TakahashiYOdaYKawaguchiKAltered expression and molecular abnormalities of cell-cycle-regulatory proteins in rhabdomyosarcomaMod Pathol20041766066915098008
- TaylorACShuLDanksMKP53 mutation and MDM2 amplification frequency in pediatric rhabdomyosarcoma tumors and cell linesMed Pediatr Oncol2000359610310918230
- HawkinsDSGuptaAARudzinskiERWhat is new in the biology and treatment of pediatric rhabdomyosarcoma?Curr Opin Pediatr201426505624326270
- HuangFGreerAHurlburtWThe mechanisms of differential sensitivity to an insulin-like growth factor-1 receptor inhibitor (BMS-536924) and rationale for combining with EGFR/HER2 inhibitorsCancer Res20096916117019117999
- RenshawJTaylorKRBishopRDual blockade of the PI3K/AKT/mTOR (AZD8055) and RAS/MEK/ERK (AZD6244) pathways synergistically inhibits rhabdomyosarcoma cell growth in vitro and in vivoClin Cancer Res2013195940595123918606
- GuentherMKGraabUFuldaSSynthetic lethal interaction between PI3K/Akt/mTOR and Ras/MEK/ERK pathway inhibition in rhabdomyosarcomaCancer Lett201333720020923684925
- ChenXStewartEShelatAATargeting oxidative stress in embryonal rhabdomyosarcomaCancer Cell20132471072424332040
- ZhangMLinardicCMKirschDGRAS and ROS in rhabdomyosarcomaCancer Cell20132468969124332036
- MerchantMSBernsteinDAmoakoMAdjuvant immunotherapy to improve outcome in high risk pediatric sarcomasClin Cancer Res2016223182319126823601
- National Cancer InstituteA pilot study of autologous T-cell transplantation with vaccine driven expansion of anti-tumor effectors after cytoreductive therapy in metastatic pediatric sarcomas Available from: https://clinicaltrials.gov/ct2/show/NCT00001566. NLM identifier: NCT00001566Accessed March 28, 2018
- National Cancer InstituteA pilot study of tumor vaccination and R-hIL-7 following standard multimodality therapy in patients with high risk pediatric solid tumors Available from: https://clinicaltrials.gov/ct2/show/NCT00923351. NLM identifier: NCT00923351Accessed March 28, 2018
- Jennerex BiotherapeuticsA phase I, open-label, dose escalation study of JX-594 (vaccinia GM-CSF/thymidine kinase-deactivated virus) administered by intratumoral injection in pediatric patients with unresectable refractory solid tumors Available from: https://clinical-trials.gov/ct2/show/NCT01169584. NLM identifier: NCT01169584Accessed March 28, 2018
- Nationwide Children’s HospitalA phase I dose escalation study of intratumoral or intravenous herpes simplex virus-1 mutant HSV1716 in patients with refractory non-central nervous system (non-CNS) solid tumors Available from: https://clinicaltrials.gov/ct2/show/NCT00931931. NLM identifier: NCT00931931Accessed March 28, 2018
- National Cancer InstituteA pilot study of tumor vaccination and R-hIL-7 following standard multimodality therapy in patients with high risk pediatric solid tumors Available from: https://clinicaltrials.gov/ct2/show/NCT00923351. NLM identifier: NCT00923351Accessed March 28, 2018
- Baylor College of MedicineTumor associated antigen (TAA)-specific cytotoxic T-lymphocytes administered to patients with solid tumors Available from: https://clinicaltrials.gov/ct2/show/NCT02239861. NLM identifier: NCT02239861Accessed March 28, 2018
- Children’s Oncology GroupA phase 2 study of IMGN901 (Lor-votuzumab Mertansine; NSC: 783609) in children with relapsed or refractory Wilms tumor, rhabdomyosarcoma, neuroblastoma, pleuropulmonary blastoma, malignant peripheral nerve sheath tumor (MPNST) and synovial sarcoma Available from: https://clinicaltrials.gov/ct2/show/NCT02452554. NLM identifier: NCT02452554Accessed March 28, 2018
- De AlavaEKawaiAHealeyJHEWS-FLI1 fusion transcript structure is an independent determinant of prognosis in Ewing’s sarcomaJ Clin Oncol199816124812559552022
- CotterillSJAhrensSPrognostic factors in Ewing’s tumor of bone: analysis of 975 patients from the European Intergroup Cooperative Ewing’s Sarcoma Study groupJ Clin Oncol2000181703108311410963639
- AngervallLEnzingerFMExtraskeletal neoplasm resembling Ewing’s sarcomaCancer19753612402511203852
- SorensenPHLessnickSLLopez-TerradaDLiuXFTricheTJDennyCTA second Ewing’s sarcoma translocation, t(21;22), fuses the EWS gene to another ETS-family transcription factor, ERGNat Genet199461461518162068
- SiebenrockKANascimentoAGRockMGComparison of soft tissue Ewing’s sarcoma and peripheral neuroectodermal tumorClin Orthop Relat Res1996288299
- TanakaMYamazakiYEwing’s sarcoma precursors are highly enriched in embryonic osteochondrogenic progenitorsJ Clin Invest20141243061307424911143
- RiggiNStamenkovicIThe biology of Ewing sarcomaCancer Lett200725411017250957
- van DoominckJAJiLSchaubBCurrent treatment protocols have eliminated the prognostic advantage of type 1 fusions in Ewing sarcoma: a report from the Children’s Oncology GroupJ Clin Oncol2010281989199420308669
- CromptonBDStewartCTaylor-WeinerAThe genomic landscape of pediatric Ewing sarcomaCancer Discov201441326134125186949
- TirodeFSurdezDMaXGenomic landscape of Ewing sarcoma defines an aggressive subtype with co-association of STAG2 and TP53 mutationsCancer Discov201441342135325223734
- MayWALessnickSLBraunBSThe Ewing’s sarcoma EWS/FLI-1 fusion gene encodes a more potent transcriptional activator and is a more powerful transforming gene than FLI-1Mol Cell Biol199313739373988246959
- JohnsonKMMahlerNRSaundRSRole for the EWS domain of EWS/FLI in binding GGAA-microsatellites required for Ewing sarcoma anchorage independent growthProc Natl Acad Sci U S A20171149870987528847958
- KinseyMSmithRLessnickSLNR0B1 is required for the oncogenic phenotype mediated by EWS/FLI in Ewing’s sarcomaMol Cancer Res2006485185917114343
- LuQZhangYMaLEWS-FLI1 positively regulates autophagy by increasing ATG4B expression in Ewing sarcoma cellsInt J Mol Med2017401217122528902354
- HeTSurdexDRantalaJKHigh-throughput RNAi screen in Ewing sarcoma cells identifies leucine rich repeats and WD repeat domain containing 1 (LRWD1) as a regulator of EWS-FLI1 driven cell viabilityGene201759613714627760381
- TannerJMBensardCWeiPEWS/FLI is a master regulator of metabolic reprogramming in Ewing sarcomaMol Cancer Res2017151517153028720588
- ChooSWangPNewburyRRobertsWYangJReactivation of TWIST1 contributes to Ewing sarcoma metastasisPediatric Blood Cancer201865e26721
- SechlerMParrishJKBirksDKJedlickaPThe histone demethylase KDM3A, and its downstream target MCAM, promote Ewing sarcoma cell migration and metastasisOncogene2017364150416028319067
- PetersHLYanYNordgrenTMCutucacheCEJoshiSSSolheimJCAmyloid precursor like protein 2 suppresses irradiation induced apoptosis in Ewing sarcoma cells and is elevated in immune evasive Ewing sarcoma cellsCancer Biol Ther20131475276023792571
- SubbiahVAndersonPLazarAJBurdettERaymondKLudwigJAEwing’s sarcoma: standard and experimental treatment optionsCurr Treat Options Oncol20091012614019533369
- KovarHBlocking the road, stopping the engine or killing the driver? Advances in targeting EWS/FLI-1 fusion in Ewing sarcoma as novel therapyExpert Opin Ther Targets2014181315132825162919
- AksnesLHBauerHCDahlAAHealth status at long-term follow-up in patients treated for extremity localized Ewing sarcoma or osteosarcoma: a Scandinavian Sarcoma Group studyPediatr Blood Cancer200953848919343771
- ParksRRaschEKManskyPJOakleyFDifferences in activities of daily living performance between long-term pediatric sarcoma survivors and a matched comparison group on standardized testingPediatr Blood Cancer20095362262819533662
- Oncternal TherapeuticsTK216 in patients with relapsed or refractory Ewing sarcoma Available from: https://clinicaltrials.gov/ct2/show/NCT02657005. NLM identifier: NCT02657005Accessed March 28, 2018
- ZöllnerSKSelvanathanSPGrahamGTInhibition of the oncogenic fusion protein EWS-FLI1 causes G2-M cell cycle arrest and enhanced vincristine sensitivity in Ewing’s sarcomaSci Signal201710eaam842928974650
- SpurnyCKailayangiriSJamitzkySProgrammed cell death ligand 1 (PD-L1) expression is not a predominant feature in Ewing sarcomasPediatr Blood Cancer201865e26719
- GroharPJGriffinLBYeungCEcteinascidin 743 interferes with the activity of EWS-FLI1 in Ewing sarcoma cellsNeoplasia20111314515321403840
- BaruchelSPappoAKrailoMA phase 2 trial of trabectedin in children with recurrent rhabdomyosarcoma, Ewing sarcoma and non-rhabdomyosarcoma soft tissue sarcomas: a report from the Children’s Oncology GroupEur J Cancer20124857958522088484
- GroharPJSegarsLEYeungCDual targeting of EWS-FLI1 activity and the associated DNA damage response with trabectedin and SN38 synergistically inhibits Ewing sarcoma cell growthClin Cancer Res2014201190120324277455
- ErkizanHVKongYMerchantMA small molecule blocking oncogenic protein EWS-FLI1 interaction with RNA helicase A inhibits growth of Ewing’s sarcomaNat Med20091575075619584866
- PrieurATirodeFCohenPDelattreOEWS/FLI-1 silencing and gene profiling of Ewing cells reveal downstream oncogenic pathways and a crucial role for repression of insulin-like growth factor binding protein 3Mol Cell Biol2004247275728315282325
- MalempatiSWeigelBIngleAMPhase I/II trial and pharmacokinetic study of cixutumumab in pediatric patients with refractory solid tumors and Ewing sarcoma: a report from the Children’s Oncology GroupJ Clin Oncol20123025626222184397
- PappoASPatelSRCrowleyJR1507, a monoclonal antibody to the insulin-like growth factor 1 receptor, in patients with recurrent or refractory Ewing sarcoma family of tumors: results of a phase II Sarcoma Alliance for Research Through Collaboration studyJ Clin Oncol2011294541454722025149
- Lamhamedi-CherradiSEMenegazBARamamoorthyVIGF-1R and mTOR blockade: novel resistance mechanisms and synergistic drug combinations for Ewing sarcomaJ Natl Cancer Inst2016108djw18227576731
- NaingALoRussoPFuSInsulin growth factor-receptor (IGF-1R) antibody cixutumumab combined with the mTOR inhibitor temsirolimus in patients with refractory Ewing’s sarcoma family tumorsClin Cancer Res2012182625263122465830
- GarnettMJEdelmanEJHeidornSJSystematic identification of genomic markers of drug sensitivity in cancer cellsNature201248357057522460902
- HeskeCMDavisMIBaumgartJTMatrix Screen identifies synergistic combination of PARP inhibitors and nicotinamide phosphoribosyltransferase (NAMPT) inhibitors in Ewing sarcomaClin Cancer Res2017237301731128899971
- ChoyEButrynskiJEHarmonDCPhase II study of olaparib in patients with refractory Ewing sarcoma following failure of standard chemotherapyBMC Cancer20141481325374341
- National Cancer InstituteNCI-COG pediatric MATCH (molecular analysis for therapy choice): a phase 2 subprotocol of olaparib in patients with tumors harboring defects in DNA damage repair genes Available from: https://clinicaltrials.gov/ct2/show/NCT03233204. NLM identifier: NCT03233204Accessed March 28, 2018
- Massachusetts General HospitalPhase I study of olaparib and temozolomide in adult patients with recurrent/metastatic Ewing’s sarcoma following failure of prior chemotherapy Available from: https://clinicaltrials.gov/ct2/show/NCT01858168. NLM identifier: NCT01858168Accessed March 28, 2018
- RajSBuiMGonzalesRLetsonDAntoniaSJImpact of PDL1 expression on clinical outcomes in subtypes of sarcomaAnn Oncol201425iv494iv510
- LeDTUramJNWangHPD-1 blockade in tumors with mismatch-repair deficiencyN Engl J Med20153722509252026028255
- Assaf-Harofeh Medical CenterA phase II single arm study assessing efficacy and safety of nivolumab plus ipilimumab in nonresectable/metastatic sarcoma and endometrial carcinoma patients with somatic deficient MMR as a selection tool Available from: https://clinicaltrials.gov/ct2/show/NCT02982486. NLM identifier: NCT02982486Accessed March 28, 2018
- von HeykingKCalzada-WackJGollnerSThe endochondral bone protein CHM1 sustains an undifferentiated, invasive phenotype, promoting lung metastasis in Ewing sarcomaMol Oncol2017111288130128319320
- ThielUPirsonSMüller-SpahnCSpecific recognition and inhibition of Ewing tumor growth by antigen-specific allo-restricted cytotoxic T cellsBr J Cancer201110494895621407224
- BlaeschkeFThielUKirschnerAHuman HLA-A 02-01/CHM1+ allo-restricted T cell receptor transgenic CD8+ T cells specifically inhibit Ewing sarcoma growth in vitro and in vivoOncotarget20167432674328027281613
- GhisoliMBarveMSchneiderRPilot Trial of FANG immu-notherapy in Ewing’s sarcomaMol Ther2015231103110925917459
- GradalisA two-part phase IIB trial of Vigil (Bi-shRNA-furin and GMCSF augmented autologous tumor cell immunotherapy) in Ewing’s sarcoma Available from: https://clinicaltrials.gov/ct2/show/NCT02511132. NLM identifier: NCT02511132Accessed March 28, 2018
- National Cancer InstituteA pilot study of tumor vaccination and R-hIL-7 following standard multimodality therapy in patients with high risk pediatric solid tumors Available from: https://clinicaltrials.gov/ct2/show/NCT00923351. NLM identifier: NCT00923351Accessed March 28, 2018
- SpurrellELFisherCThomasJMJudsonIRPrognostic factors in advanced synovial sarcoma: an analysis of 104 patients treated at the Royal Marsden HospitalAnn Oncol20051643744415653701
- NielsenTOPoulinNMLadanyiMSynovial sarcoma: recent discoveries as a roadmap to new avenues for therapyCancer Discov2015512413425614489
- HerzogCEOverview of sarcomas in the adolescent and young adult populationJ Pediatr Hematol Oncol200527215815838394
- LadanyiMAntonescuCRLeungDHImpact of SYT-SSX fusion type on the clinical behavior of synovial sarcoma: a multi-institutional retrospective study of 243 patientsCancer Res20026213514011782370
- SultanIRodriguez-GalindoCSaabRYasirSCasanovaMFerrariAComparing children and adults with synovial sarcoma in the Surveillance, Epidemiology, and End Results program, 1983 to 2005:an analysis of 1268 patientsCancer20091153537354719514087
- FisherCSynovial sarcomaAnn Diagn Pathol199824014219930576
- ClarkJRocquesPJCrewAJIdentification of novel genes, SYT and SSX, involved in the t(X;18)(p11.2;q11.2) translocation found in human synovial sarcomaNat Genet199475025087951320
- NagayamaSKatagiriTTsunodaTGenome-wide analysis of gene expression in synovial sarcomas using a cDNA microarrayCancer Res2002625859586612384549
- WeissSWSobinLHHistological Typing of Soft Tissue Tumours2nd edHeidelbergSpringer1994
- ClarkJRocquesPJCrewAJIdentification of novel genes, SYT and SSX, involved in the t(X;18)(p11.2;q11.2) translocation found in human synovial sarcomaNat Genet199475025087951320
- SkyttingBNilssonGBrodinBA novel fusion gene, SYT-SSX4, in synovial sarcomaJ Natl Cancer Inst19999197497510359553
- KawaiAWoodruffJHealeyJHBrennanMFAntonescuCRLadanyiMSYT-SSX gene fusion as a determinant of morphology and prognosis in synovial sarcomaN Engl J Med19983381531609428816
- AntonescuCRKawaiALeungDHStrong association of SYT-SSX fusion type and morphologic epithelial differentiation in synovial sarcomaDiagn Mol Pathol200091810718206
- NakaqawaYNumotoKYoshidaAChromosomal and genetic imbalances in synovial sarcoma detected by conventional and microarray comparative genomic hybridizationJ Cancer Res Clin Oncol200613244445016557383
- SzymanskaJSerraMSkyttingBGenetic imbalances in 67 synovial sarcomas evaluated by comparative genomic hybridizationGenes Chromosomes Cancer1998232132199790501
- AllanderSVIlleiPBChenYExpression profiling synovial sarcoma by cDNA microarrays: association of ERBB2, IGFBP2, and ELF3 with epithelial differentiationAm J Pathol20021611587159512414507
- SaitoTOdaYSakamotoAPrognostic value of the preserved expression of the E-cadherin and catenin families of adhesion molecules and of B-catenin mutations in synovial sarcomaJ Pathol200019234235011054718
- NielsenTHsuFDO’ConnellJTissue microarray validation of epidermal growth factor receptor and SALL2 in synovial sarcoma with comparison to tumors of similar histologyAm J Pathol20031631449145614507652
- TerryJSaitoTSubramanianSTLE1 as a diagnostic immunohistochemical marker for synovial sarcoma emerging from gene expression profiling studiesAm J Surg Pathol20073124024617255769
- Washington University School of MedicineA phase I/II clinical trial of dose-escalating DHEA in synovial sarcoma patients Available from: https://clinicaltrials.gov/ct2/show/NCT02683148. NLM identifier: NCT02683148Accessed March 28, 2018
- EpizymeA phase 1 study of the EZH2 inhibitor tazemetostat in pediatric subjects with relapsed or refractory INI1-negative tumors or synovial sarcoma Available from: https://clinicaltrials.gov/ct2/show/NCT02601937. NLM identifier: NCT02601937Accessed March 28, 2018
- EpizymeA phase II, multicenter study of the EZH2 inhibitor tazemetostat in adult subjects with INI1-negative tumors or relapsed/refractory synovial sarcoma Available from: https://clinicaltrials.gov/ct2/show/NCT02601950. NLM identifier: NCT02601950Accessed March 28, 2018
- Advenchen LaboratoriesA phase III study of AL3818 (anlotinib) hydrochloride monotherapy in subjects with metastatic or advanced alveolar soft part sarcoma, leiomyosarcoma, and synovial sarcoma Available from: https://clinicaltrials.gov/ct2/show/NCT03016819. NLM identifier: NCT03016819Accessed March 28, 2018
- National Cancer InstituteA phase 1B/2 study of imatinib in combination with everolimus in synovial sarcoma Available from: https://clinicaltrials.gov/ct2/show/NCT01281865. NLM identifier: NCT01281865Accessed March 28, 2018
- Memorial Sloan Kettering Cancer CenterPhase II trial of sorafenib and dacarbazine in soft tissue sarcoma Available from: https://clinicaltrials.gov/ct2/show/NCT00837148. NLM identifier: NCT00837148Accessed March 28, 2018
- KawaguchiSWadaTIdaKPhase I vaccination trial of SYT-SSX junction peptide in patients with disseminated synovial sarcomaJ Transl Med20053115647119
- SuminoeAMatsuzakiAHattoriHKogaYHaraTImmunotherapy with autologous dendritic cells and tumor antigens for children with refractory malignant solid tumorsPediatr Transplant20091374675319067917
- KawaguchiSTsukaharaTIdaKSYT-SSX breakpoint peptide vaccines in patients with synovial sarcoma: a study from the Japanese Musculoskeletal Oncology GroupCancer Sci20121031625163022726592
- JungbluthAAAntonescuCRBusamKJMonophasic and biphasic synovial sarcomas abundantly express cancer/testis antigen NY-ESO-1 but not MAGE-A1 or CT7Int J Cancer20019425225611668506
- RobbinsPFMorganRAFeldmanSATumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1J Clin Oncol201129911924
- JagerENagataYGnjaticSMonitoring CD8 T cell responses to NY-ESO-1: correlation of humoral and cellular immune responsesProc Natl Acad Sci U S A2000974760476510781081
- StockertEJagerEChenYTA survey of the humoral immune response of cancer patients to a panel of human tumor antigensJ Exp Med1998187134913549547346
- van den EyndeBJvan der BruggenPT cell defined tumor antigensCurr Opin Immunol199796846939368778
- AdaptimmuneA pilot study of genetically engineered NY-ESO-1 specific NY-ESO-1 C259T in HLA-A2+ patients with synovial sarcoma Available from: https://clinicaltrials.gov/ct2/show/NCT01343043. NLM identifier: NCT01343043Accessed March 28, 2018
- National Cancer InstitutePhase II study of metastatic cancer that expresses NY-ESO-1 using lymphodepleting conditioning followed by infusion of anti-NY-ESO-1 murine TCR-gene engineered lymphocytes Available from: https://clinicaltrials.gov/ct2/show/NCT01967823. NLM identifier: NCT01967823Accessed March 28, 2018
- Takara BioMulti-center phase I/II study of NY-ESO-1 T cell receptor gene transferred T lymphocytes in patients with synovial sarcoma Available from: https://clinicaltrials.gov/ct2/show/NCT03250325. NLM identifier: NCT03250325Accessed March 28, 2018
- Immune DesignA randomized, open-label, phase II trial of CMB305 (sequentially administered LV305 and G305) and atezolizumab in patients with locally advanced, relapsed, or metastatic sarcoma expressing NY-ESO-1 Available from: https://clinicaltrials.gov/ct2/show/NCT02609984. NLM identifier: NCT02609984Accessed March 28, 2018
- Petrov Research Institute of OncologyNon-randomized single-center study phase II evaluating the efficacy and toxicity of autologous dendritic cell vaccine loaded with allogenic tumor lysate expression of cancer testis antigens in patients with soft tissue sarcoma Available from: https://clinicaltrials.gov/ct2/show/NCT01883518. NLM identifier: NCT01883518Accessed March 28, 2018
- ZhangPPollockREEpigenetic regulators: new therapeutic targets for soft tissue sarcomaCancer Cell Microenviron20141e19126078988
- KawaguchiKOdaYSaitoTDNA hypermethylation status of multiple genes in soft tissue sarcomasMod Pathol200614106114
- LubienieckaJMde BruijnDRSuLHistone deacetylase inhibitors reverse SS18-SSX-mediated polycomb silencing of the tumor suppressor early growth response 1 in synovial sarcomaCancer Res2008684303431018519690
- WangHGarzonRSunHNF-κB-YY1-miR-29 regulatory circuitry in skeletal myogenesis and rhabdomyosarcomaCancer Cell20081436938118977326