
Abstract
Introduction
Targeted therapies using epidermal growth factor receptor-tyrosine kinase inhibitors (EGFR-TKIs) have revolutionized the treatment of non-small cell lung cancer (NSCLC) patients harboring EGFR mutations, leading to the approval of gefitinib and erlotinib as standard first-line clinical treatment. Inevitably, a considerable proportion of patients develop resistance to EGFR-TKIs due to the acquisition of secondary mutations within EGFR. Therefore, alternative strategies to target NSCLC are desperately needed.
Materials and methods
In this study, we have evaluated the effect of a reactive oxygen species (ROS)-inducing agent WZ35, a mono-carbonyl analog of curcumin, to target an inherent biological property of cancer cells, increased oxidative stress. To study whether WZ35 can inhibit NSCLC tumorigenesis, we used gefitinib- and erlotinib-resistant cell line H1975.
Results
In this study, we show that WZ35 treatment dramatically decreases cell viability and induces apoptosis in H1975 cells through the generation of ROS. We also found that the antitumor activity of WZ35 involved ROS-mediated activation of the endoplasmic reticulum stress pathway and mitochondrial dysfunction. Furthermore, WZ35 significantly inhibited H1975 xenograft tumor growth through the inhibition of cell proliferation and induction of apoptosis.
Discussion
These findings show that WZ35 may be a promising candidate for the treatment of EGFR-TKI-resistant NSCLC.
Introduction
Lung cancer is one of the leading causes of death in men and women.Citation1 It can be broadly divided into small cell lung carcinoma and non-small cell lung cancer (NSCLC), with NSCLC accounting for nearly 85% of all cases.Citation2,Citation3 If diagnosed early, the primary treatment for NSCLC is surgical resection. However, most cases are diagnosed at a later stage and are treated with chemotherapy with an overall survival rate of <2 years.Citation4 Epidermal growth factor receptor-tyrosine kinase inhibitors (EGFR-TKIs) provided initial hope for patients with NSCLC harboring EGFR activating mutations.Citation5–Citation7 Patients who initially responded well to EGFR-TKIs gefitinib and erlotinib had mutations in the kinase domain of EGFR. The two most common EGFR mutations were found to be short in-frame deletions of exon 19 and a point mutation in exon 21 which results in L858R.Citation8–Citation10 However, majority of these patients acquire resistance through a secondary mutation resulting in T790M.Citation11,Citation12 Most recently, third-generation mutant-selective EGFR inhibitors, such as AZD9291 and CO-1686, emerged to inhibit tumor growth in patients with T790M-positive NSCLC. Again, acquired resistance to and failure of the third-generation TKIs is a major concern.Citation13,Citation14 Therefore, alternative strategies are desperately needed for the treatment of patients with EGFR T790M-positive NSCLC.
Besides targeting EGFR mutations in NSCLC, the inherent biological differences between cancer cells and normal cells may be of value. Evidence from recent studies show that cancer cells, unlike normal cells, are under increased oxidative stress.Citation15,Citation16 Increased generation of reactive oxygen species (ROS) and resulting oxidative stress is believed to facilitate oncogenic transformations and metabolic activity of cancer cells. As ROS can inflict cellular damage by negatively modifying lipids, proteins, and DNA, increasing ROS to toxic levels in cancer cells provide an interesting target for treatment. A previous study reporting arsenic trioxide-induced increases in expression and activity of caspases in leukemia cells has brought forward the implication of ROS elevation in cancer environment,Citation17,Citation18 the strategy of increasing ROS in cancer is again gaining traction. Our laboratory focused on evaluating the antitumor effects of ROS-generating agents both in vitro and in vivo.Citation19–Citation21 In these studies, we showed that a novel analog of curcumin, WZ35, induces ROS generation and subsequent cell apoptosis in pancreatic, colon, and gastric cancer. However, the antitumor effects of WZ35 against NSCLC are still unknown.
In this study, we investigated the effect of WZ35 on the gefitinib- and erlotinib-resistant H1975 cell line, which harbors L858R and T790M mutations.Citation11 Based on our previous work in pancreatic and colorectal cancer, we hypothesized that WZ35 will induce ROS generation in H1975 and inhibit cancer growth. Indeed, we show that WZ35 induced apoptosis in H1975 cells by increasing ROS levels. Elevated ROS then produces catastrophic cellular responses illustrated by the activation of endoplasmic reticulum (ER) stress and mitochondrial dysfunction. We also show that WZ35 inhibited the growth of gefitinib- and erlotinib-resistant H1975 tumors in a xenograft model by inhibiting proliferation and inducing apoptosis.
Materials and methods
Cell culture and reagents
Human NSCLC cell line H1975 was purchased from the Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences (Shanghai, People’s Republic of China). H1975 is a gefitinib-resistant line and harbors L858R and T790M mutations in EGFR. Cells were cultured in RPMI 1640 medium (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% heat-inactivated fetal bovine serum (Hyclone, Logan, UT, USA), 100 units/mL penicillin, and 100 µg/mL streptomycin.
WZ35 was synthesized in our laboratory to a purity of 98.8% as described previously.Citation21 WZ35 was dissolved in DMSO for in vitro experiments and in PEG-castor oil-physiological saline with a volume ratio of 8:1:11 for in vivo experiments. Gefitinib/erlotinib (Aladdin, Shanghai, China) was suspended in DMSO and stored in volumes of 1 mL at −20°C, and NAC was purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Fluorescein isothiocyanate (FITC)-Annexin V apoptosis detection kit and PI were purchased from BD Diagnostics (Franklin Lakes, NJ, USA). Antibodies against cleaved (Cle)-PARP, Bcl2 associated protein x (Bax), B-cell lymphoma 2 (Bcl-2), MDM-2, cyclin B1, CDC2, caspase-3, cytochrome c, TMRM, DIOC6(3), Ki67, GAPDH, and horseradish peroxidase (HRP)-conjugated secondary antibodies were purchased from Santa Cruz Biotechnology Inc. (Dallas, TX, USA). Antibodies against ATF4, p-eIF2α, CCAAT/enhancer-binding protein homologous protein (CHOP), and Cle-caspase 3 were purchased from Cell Signaling Technology (Danvers, MA, USA). DCFH-DA and JC-1 were purchased from Beyotime Biotech (Nantong, China).
Cell viability assay
H1975 cells were plated in 96-well tissue culture plates at a density of 5000 per well. Cells were allowed to attach overnight. The following day, cells were treated with gefitinib, erlotinib, and WZ35 at various concentrations for 24 h. Cell viability was then measured through MTT assay. Gefitinib, erlotinib, and WZ35 were dissolved in DMSO and then diluted in RPMI 1640 medium to the desired final concentration.
Flow cytometry for apoptosis and cell cycle analysis
For the determination of apoptosis, cells were plated in 60-mm dishes and allowed to attach overnight. Cells were then treated with WZ35 at various concentrations for 24 h. Following treatments, cells were harvested and resuspended in binding buffer at a concentration of 106 cells/mL. Apoptosis was determined by double staining cells with FITC-Annexin V and PI in binding buffer for 30 min and analysis by using FACSCalibur flow cytometer (BD Biosciences, San Jose, CA, USA). For cell cycle analysis, cells were treated as indicated and harvested as indicated. We then fixed the cells in 75% ethanol at 4°C overnight. After washing the cells, PI staining was carried out in solution containing RNase for 30 min. Data for apoptosis and cell cycle distribution were analyzed by using FlowJo7.6 software.
Determination of cellular ROS
Cellular ROS contents were measured by flow cytometry. Cells were plated in 60-mm dishes at a density of 5×105 cells and allowed to attach overnight. Cells were then exposed to different concentrations of WZ35 for 30 min. In some experiments, cells were pretreated with 5 mM NAC for 2 h prior to WZ35 exposure. Following treatments, cells were stained with 10 µM DCFH-DA at 37°C for 30 min. Fluorescence was measured by using FACSCalibur flow cytometer (BD Biosciences).
Electron microscopy
Following treatments, cells were collected and fixed in phosphate buffer (pH 7.4) containing 2.5% glutaraldehyde overnight at 4°C. Cells were then postfixed in 1% OsO4 at room temperature for 60 min, stained with 1% uranyl acetate, dehydrated through graded acetone solutions, and embedded in Epon. Areas containing cells were block mounted and cut into 70-nm sections and examined with an electron microscope (H-7500; Hitachi Ltd., Tokyo, Japan).
Western blot analysis
Cells or tumor tissues were homogenized in protein lysis buffer. Samples were centrifuged at 12,000 rpm for 10 min at 4°C. Protein concentrations were determined by using the Bradford protein assay (Bio-Rad Laboratories Inc., Hercules, CA, USA). Protein samples were separated by sodium dodecyl sulfate-polyacryl-amide gel electrophoresis and electro-transferred to poly-vinylidene difluoride transfer membranes. The blots were blocked for 2 h at room temperature with fresh 5% nonfat milk in tris-buffered saline Tween-20 (TBST) and then incubated with specific primary antibody in TBST overnight at 4°C. Following three washes with TBST, the blots were incubated with HRP-conjugated secondary antibodies for 1 h, and the bands were visualized by using ECL kit (Bio-Rad Laboratories Inc.). The density of the bands was analyzed by using Image J (National Institute of Health, Bethesda, MD, USA).
Immunofluorescence staining
Cells were grown in 35-mm cell culture dish with glass bottom (NEST, Wuxi, China), fixed with 4% paraformaldehyde (Sigma-Aldrich Co.) for 15 min at room temperature, and then permeabilized with 0.25% Triton X-100-PBS for 10 min at 4°C. Blocking was performed with 1% bovine serum albumin (BSA)-PBS. Cells were then incubated with indicated primary antibody (1:200) diluted in 1% BSA-PBS at 4°C overnight. Fluorophore-conjugated second antibody (1:200) diluted in the same buffer was added for 1 h. Cells were counterstained with 4′,6-diamidino-2-phenylindole. Fluorescence was detected using DMI6000 microscope (Leica Microsystems, Wetzlar, Germany).
Transient transfection of siRNA
siRNA duplexes were purchased from Thermo Fisher Scientific. CHOP siRNA was custom designed (NCBI accession no. NM_004083, sense sequence 5′-GAGCUCUGAUUGACCGAAUGGUGAA-3′). Negative Universal Control siRNA was also purchased from Thermo Fisher Scientific. To transfect cells, H1975 cells were plated at a density of 3×105/well in a 6-well plate and cultured for 24 h. siRNA against CHOP (100 nM) or control siRNA was transfected by using lipofectamine 2000 (Thermo Fisher Scientific). Cells were further cultured for 48 h before harvesting for the detection of CHOP protein by Western blotting.
Evaluation of mitochondrial membrane potential (Δψm)
The effects of WZ35 on Δψm was examined by using JC-1, DiOC6(3), or TMRM. We treated cells with WZ35 for 14 h and then stained with JC-1, DiOC6(3), or TMRM for 30 minutes. Images were acquired by using a Nikon fluorescence microscope (Nikon Corporation, Tokyo, Japan).
H1975 xenografts
All animal experiments complied with the Wenzhou Medical University’s Policy on the Care and Use of Laboratory Animals. Protocols for animal studies were approved by the Wenzhou Medical College Animal Policy and Welfare Committee (Approved documents: 2012/APWC/0216). Five-week-old athymic BALB/cA nu/nu female mice (18–22 g) were purchased from Vital River Laboratories (Beijing, China). Animals were housed at a constant room temperature with a 12:12 h light/dark cycle and fed a standard rodent diet and given water ad lib. H1975 cells were injected subcutaneously into the right flank at 1×107 cells in 150 µL of PBS per mouse. When tumors reached a volume of 50–100 mm3, tumor-bearing mice were treated with WZ35 by intra-peritoneal injections twice a day (2.5 or 5 mg/kg/d). WZ35 was dissolved in PEG-castor oil-physiological saline with a volume ratio of 8:1:11. Tumor volumes were determined by measuring length (l) and width (w) and calculating volume (V =0.5×l×wCitation2) at the indicated time points. At the end of treatment, the animals were sacrificed, and the tumors were removed and weighed for use in histology and proteins expression studies.
Immunohistochemistry
The harvested tumor tissues were fixed in 10% formalin at room temperature, processed, and embedded in paraffin. Paraffin-embedded tissues were sectioned at 5 µm thickness. Tissue sections were deparaffinized, rehydrated, and incubated with primarily Ki-67 antibody. HRP-conjugated secondary antibody and diaminobenzidine were used for detection. Images were obtained with Image-Pro Plus 6.0 (Media Cybernetics, Inc., Bethesda, MD, USA).
Statistical analysis
All experiments were performed at least three times with three replicates (n=3). Data are expressed as mean ± SEM. All statistical analyses were performed by using Graph-Pad Pro Prism 5.0 (GraphPad Software, Inc., La Jolla, CA, USA). Student’s t-test and two-way analysis of variance were employed to analyze the differences between sets of data. A p-value <0.05 was considered statistically significant.
Results
WZ35 induces apoptotic cell death in H1975 lung cancer cells
We first assessed whether WZ35 () alters the viability of H1975 cells. We treated cells with H1975 as well as gefitinib and erlotinib and measured viability through the MTT assay. Based on the dose–response curve, the IC50 value of WZ35 was calculated to be 1.17 µM, whereas those of gefitinib and erlotinib were found to be >20 µM (). This clearly indicates that WZ35 is far more competent to inhibit cell growth than the conventional EGFR-TKIs.
Figure 1 WZ35 inhibits cell growth and induces apoptosis in H1975 cells. (A) Chemical structures of compound WZ35. (B) The effects of WZ35, erlotinib, and gefitinib on the viability of H1975 cells. H1975 cells were incubated with increasing concentrations of WZ35, erlotinib, and gefitinib (0.625–20 μM) for 24 h. Cell viability was determined by MTT assay, and the IC50 values were calculated. (C) Apoptosis in H1975 cells was determined by flow cytometry after treating cells with WZ35 for 24 h. (D) Quantification of data presented in panel D (*P<0.05, **P<0.01, and ***P<0.001 compared with DMSO control). (E–I) H1975 cells were treated with WZ35 for 20 h. Cell lysates were subjected to Western blot analysis for apoptosis-related proteins, cleaved-PARP (E), Bax (F), Bcl-2 (G), Pro-caspase 3 (H), and Cle-caspase 3 (I). GAPDH was used as internal control.
Abbreviations: DMSO, dimethyl sulfoxide; PI, propidium iodide; FITC, fluorescein isothiocyanate; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide.
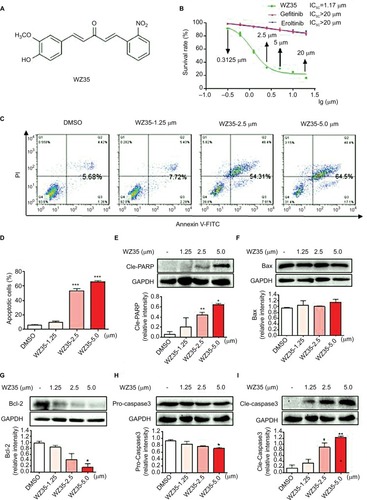
To decipher whether the remarkable reduction of viability induced by WZ35 treatment was ascribed to increased apoptotic activity, we performed flow cytometric analysis of Annexin V/propidium iodide (PI). Our flow cytometric analysis indicated that WZ35 provoked apoptosis in a dose-dependent manner (), which is consistent with the concomitant elevation of cleaved poly ADP-ribose polymerase (PARP) and Cle-caspase 3 () and decreases in Bcl-2 and Pro-caspase 3 (). There was no significant change in the expression of Bax after WZ35 treatment ().
WZ35 causes G2/M cell cycle arrest in H1975 cells
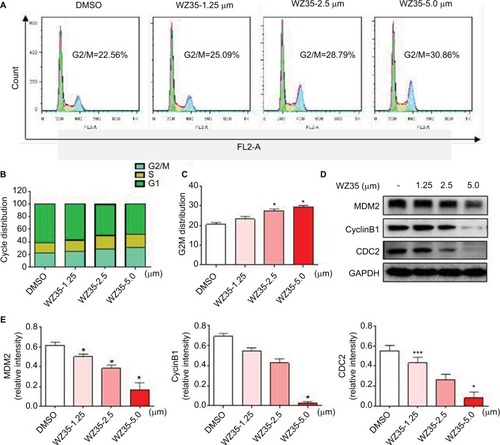
We next determined the effect of WZ35 on the growth of H1975 cells. We treated the cells with various concentrations of WZ35 for 24 h and then assessed cell cycle phase distribution. Our results show that WZ35 induced G2/M cell cycle arrest in H1975 (). G2/M is an important DNA damage checkpoint and involves a number of regulatory proteins including murine double minute 2 (MDM2), cyclin B1, and cyclin-dependent kinase 1 (CDC2). We assessed the level of these proteins in H1975 cells exposed to WZ35 and show that WZ35 caused a dose-dependent decrease in the levels of MDM-2, cyclin B1, and CDC2 (). Together, these data reveal that WZ35 induced reduction of viability of H1975 via G2/M phase arrest and induction of apoptosis.
Figure 2 WZ35 induces cell cycle arrest in H1975 cells. (A) Induction of cell cycle arrest in H1975 cells was determined by PI staining and flow cytometry, after treating cells with WZ35 for 18 h. (B) The distribution of cell cycle phase in cells treated with WZ35. (C) Representative histograms from flow cytometry analysis in the three TNBC cells treated with osthole. (*P<0.05 compared with DMSO control). (D) Expression of cell cycle-associated proteins showing decreased levels of MDM2, cyclin B1, and CDC2 following treatment of cells with WZ35 for 18 h. GAPDH was used as internal control. (E) Western blot results from (D) was calculated and represented as the percent of control. (*P<0.05 and ***P<0.001 compared with DMSO control).
Abbreviations: DMSO, dimethyl sulfoxide; PI, propidium iodide; MDM2, murine double minute; CDC2, cyclin-dependent kinase 1.
Elevated ROS produces WZ35-induced apoptosis
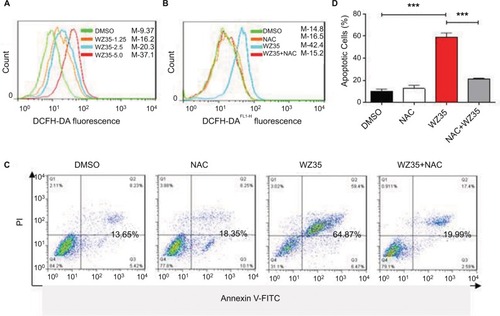
We have recently shown that WZ35 induces apoptosis in pancreatic and gastric cancer cells through the generation of ROS.Citation20,Citation21 Next, we determined the apoptotic effects and mechanism of WZ35 in H1975 cells. In concordant with our previous experiments, we found that WZ35 induced apoptosis in H1975 cells by inducing oxidative stress. To measure ROS levels, we used dichloro-dihydro-fluorescein diacetate (DCFH-DA) after exposing the cells to WZ35 at various concentrations for 30 min. Increased fluorescence, indicative of increased ROS levels, was seen in cells treated with WZ35 (; M shows mean fluorescence intensity). To validate this assay, we pretreated cells with 5 mM N-acetylcysteine (NAC) for 2 h before challenging the cells with WZ35. NAC is an antioxidant and readily quenches ROS levels. As shown in , NAC pretreatment prevented WZ35-induced apoptosis. These results revealed that ROS was mediating apoptosis in WZ35-challenged cells. We show that WZ35-induced cell apoptosis () was completed prevented by pretreating the cells with NAC. These findings demonstrate that WZ35 induces ROS in H1975 cells and leads to ROS-dependent apoptosis.
Figure 3 WZ35 increases cell apoptosis through ROS generation. (A) Intracellular ROS generation induced by increasing doses of WZ35. ROS was measured by staining cells with DCFH-DA after 24 h of WZ35 challenge. Increase in fluorescence is indicative of increased ROS levels. (B) H1975 cells were pretreated with 5 mM NAC for 2 h before exposing the cells to 5 μM WZ35. ROS levels were then measured as in panel A. (C) NAC pretreatment prevents WZ35-induced apoptosis in H1975 cells as measured by Annexin-V/PI staining. (D) Quantification of data presented in panel C. Data acquired from three independent experiments. ***P<0.001.
Abbreviations: ROS, reactive oxygen species; DCFH-DA, dichloro-dihydro-fluorescein diacetate; NAC, N-acetylcysteine; PI, propidium iodide; DMSO, dimethyl sulfoxide; FITC, fluorescein isothiocyanate.
WZ35 activates ER stress signaling by promoting the accumulation of ROS in H1975 cells
Increased ROS has been shown to elicit the unfolded protein response (UPR) and the activation of the ER stress pathway leading to apoptosis.Citation22,Citation23 We have also shown that WZ35 activates the ER stress pathway in cells.Citation19,Citation20 Hence, we investigated the ER stress in H1975 cells treated with WZ35. We first analyzed the morphology of ER by electron microscopy. Swollen ER (red arrows, ) was shown in WZ35-treated cells but not in dimethyl sulfoxide (DMSO) control. Furthermore, combination of treatment with WZ35 and NAC led to comparable ER morphology to the DMSO control, suggesting that ROS was inducing ER stress in H1975 cells. We confirmed these findings by analyzing important proteins in the ER stress pathway. Various stress signals are known to induce UPR through the phosphorylation of eukaryotic initiation factor 2α (eIF2α).Citation24 One protein that is spared from this global repression is activating transcription factor 4 (ATF4). ATF4 translation increases and subsequently causes the induction of C/EBP-homologous protein (CHOP) and promotion of ER stress-induced apoptosis.Citation25 Based on these studies, we assessed the protein levels of phosphorylated eIF2α (p-eIF2α), ATF4, and CHOP as readouts of the ER stress pathway activation in H1975 cells.Citation26,Citation27 WZ35 elevated significantly the protein contents of p-eIF2α, ATF4, and CHOP in both time- and dose-dependent manners (), whereas these increases were blocked by pretreatment with NAC (). CHOP immunoreactivity was increased only by WZ35 but not in conjunction with NAC ( and ). These results clearly show that ROS generated by WZ35 activated the ER stress pathway in H1975 cells.
Figure 4 WZ35-induced H1975 apoptosis involves ROS-dependent ER stress. (A) EM evaluation of ER in H1975. H1975 cells were exposed to 5 μM WZ35 for 6 h, with or without pretreatment with 5 mM NAC for 2 h. The morphology of ER was examined with an electron microscope (×30,000) showing ER swelling (red arrows) in H1975 with WZ35 exposure. (B) H1975 cells were treated with 5 μM WZ35 for different time periods, and the protein levels of p-eIF2α, ATF4, and CHOP were determined by Western blotting. (C) H1975 cells were treated with WZ35 at different concentrations for 6 or 12 h. ER stress-associated proteins were then detected. (D) H1975 cells were pretreated with 5 mM NAC for 2 h before exposure to 5 μM WZ35 for 6 or 12 h. Same analysis as in panels B and C was performed. GAPDH was used as internal control. Data presented are representative of three independent experiments. (E) Immunostaining of H1975 for CHOP (red). Cells were pretreated with 5 mM NAC for 2 h before exposure to 5 μM WZ35 for 12 h. Cells were counterstained with DAPI (blue). (F) H1975 cells transfected with CHOP siRNA or negative control siRNA and then treated with 5 μM WZ35 for 12 h. CHOP expression in H1975 cells was determined by Western blotting. (G) Percentage of cell apoptosis following CHOP siRNA transfection and treatment of cells with WZ35 for 24 h. Apoptotic cells were measured by Annexin-V/PI staining.
Abbreviations: ROS, reactive oxygen species; EM, electron microscopy; ER, endoplasmic reticulum; NAC, N-acetylcysteine; DMSO, dimethyl sulfoxide; siRNA, small interfering RNA; PI, propidium iodide; FITC, fluorescein isothiocyanate; CHOP, C/EBP-homologous protein; ATF4, activating transcription factor 4; p-eIF2α, phosphorylated eukaryotic initiation factor α; DAPI, 4′,6-diamidino-2-phenylindole.
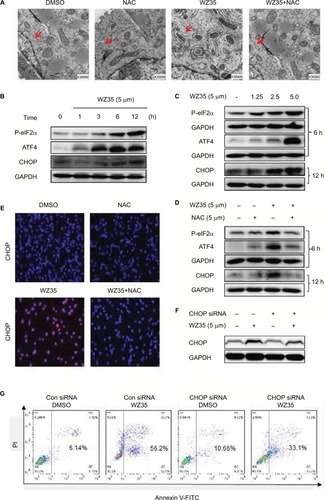
Overexpression of CHOP has been reported to lead to cell cycle arrest and/or apoptosis in a number of cell types.Citation28–Citation30 We next proposed to confirm the key role of ER stress in the induction of H1975 cell apoptosis exposed to WZ35. To test this, we knocked down CHOP by small interfering RNA (siRNA) (). Following knockdown, we exposed the cells to WZ35 and assessed the level of apoptotic cell death by Annexin V/PI staining. Our results show that CHOP knockdown significantly decreased WZ35-induced apoptotic cell death in H1975 cells (). These results show that WZ35-induced cell apoptosis is, at least partly, mediated by the activation of the ER stress pathway and induction of CHOP.
WZ35 induces ROS-dependent mitochondrial dysfunction in H1975 cells
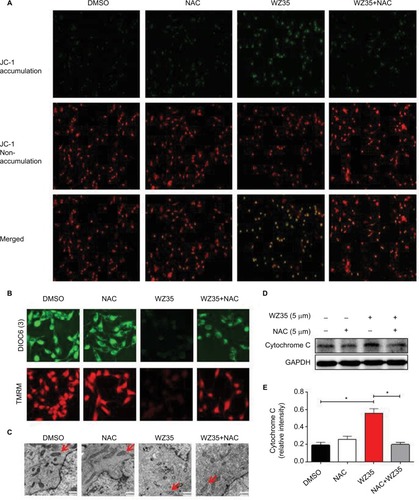
Previously, we found that ROS elevation was an important mediator of the beneficial effects of WZ35 in terms of causing ER stress activation and inhibiting cancer cell growth. ER is a crucial subcellular organelle; along with mitochondria, it plays a fundamental role in the intracellular calcium store and synthesis of cholesterol, steroids, and numerous lipid constituents.Citation31,Citation32 Furthermore, the ER–mitochondria interaction supports communication between the two organelles, including the exchange of Ca2+, which controls ER chaperone protein, mitochondrial ATP production, and apoptosis.Citation33 A large body of evidence indicates that therapy-induced oxidative stress may activate ER stress and mitochondria-related apoptosis.Citation34 Loss of mitochondrial membrane potential (Δψ m) is considered as a hallmark of mitochondrial dysfunction. This loss is catastrophic for cells and leads to the release of cytochrome c into the cytosol and subsequent induction of apoptosis. We assessed whether mitochondrial dysfunction is also present in WZ35-treated H1975 cells. We used three fluorescence dyes to examine mitochondrial membrane potential. These include JC-1, DiOC6(3), and TMRM. JC-1 is a cell permeable dye which exhibits potential-dependent accumulation in mitochondria, indicated by a fluorescence emission shift from green (529 nm) to red (590 nm). DiOC6(3) is also a cell-permeable fluorescence dye that is selective for the mitochondria of live cells. Finally, TMRM accumulates in negatively charged polarized mitochondria and fluoresces. When mitochondrial membrane potential collapses, TMRM reagent is dispersed through the cell cytosol and fluorescence levels drop. All three dyes provide assessment of mitochondrial membrane potential. Our results show that the integrity of mitochondrial membranes was compromised when cells were exposed to WZ35 ( and ). In terms of JC-1, we noted a change in fluorescence from red to green. In addition, overall fluorescence from DiOC6(3) and TMRM dispersed and dropped in the same condition. No noticeable changes, compared to control cells, were observed when the cells were pretreated with NAC before WZ35 exposure. These findings show ROS-mediated collapsing mitochondrial membrane potential in H1975 cells exposed to WZ35. We confirmed mitochondrial deficits by examining structural changes through electron microscopy. Our findings indicate swollen mitochondria and disrupted cristae when cells were exposed to WZ35 (). These morphological changes were not observed if the cells were pretreated with NAC. The mitochondrial disruptions seen in cells exposed to WZ35 were associated with increased cytochrome c protein in H1975 cells ().
Figure 5 WZ35 induces ROS-dependent mitochondrial dysfunction in H1975 cells. (A) Treatment of cells with 5 μM WZ35 decreased mitochondrial membrane potential (Δψm). Cells were exposed to WZ35 for 14 h, with or without 5 mM NAC pretreatment, and then stained with JC-1 (JC-1 accumulation in the mitochondria appear red, non-accumulating JC-1 as a monomer appear green; scale bar = 20 μM). (B) Mitochondrial membrane potential changes as determined by DiOC6(3) dye (green) and TMRM dye (red). Cells were treated as in panel A. (C) Effect of NAC on WZ35-induced changes in mitochondrial morphology in H1975. Cells were treated as in panel A and harvested for EM analysis. Swollen mitochondria with disrupted cristae can be seen in cells exposed to WZ35. (D) Levels of cytochrome c in H1975 cells exposed to WZ35 for 14 h, with or without 5 mM NAC pretreatment for 2 h. GAPDH was used as internal control. Data presented are representative of three independent experiments. (E) Densitometric quantification of cytochrome c Western blot data (*P<0.05 compared to DMSO control).
Abbreviations: ROS, reactive oxygen species; NAC, N-acetylcysteine; DMSO, dimethyl sulfoxide; EM, electron microscopy; TMRM, tetramethylrhodamine, methyl ester.
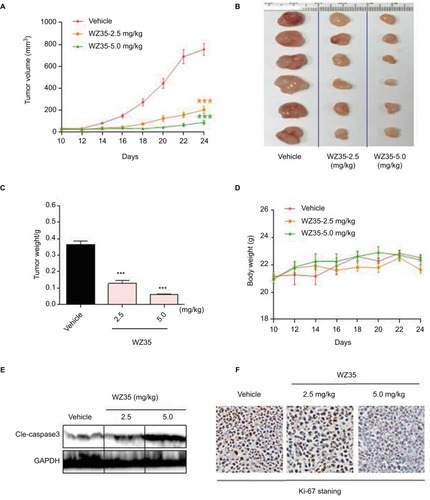
WZ35 inhibits H1975 xenograft growth in vivo
Our last objective was to determine whether WZ35 induces apoptosis and prevents the growth of gefitinib- and erlotinib-resistant H1975 lung cancer in a xenograft model. H1975 xenograft model was developed in nude mice. The tumor-bearing mice were treated with WZ35 by intraperitoneal injections at two different doses: 2.5 or 5 mg/kg. As shown in , treatment with WZ35 resulted in significant reductions in both tumor volume and weight compared to the vehicle group. No significant changes in body weights were noted in any of the experimental groups (). We then assessed cell apoptosis by analyzing active caspase 3 levels in lysates prepared from the tumors. Our results show dramatically high cleaved caspase 3 levels in tumors removed from mice treated with 5 mg/kg WZ35 (). These results indicate active apoptotic cell death in tumors treated with WZ35. In addition, H1975 tumors (vehicle treated) showed robust ki-67 immunoreactivity indicating cellular proliferation (). The level of ki-67 immunoreactivity was reduced in tumors from mice receiving 2.5 mg/kg WZ35, and only scattered proliferating cells were noted in mice that received 5 mg/kg WZ35. Collectively, these in vivo studies revealed that WZ35 prevents H1975 tumor growth by inhibiting proliferation and inducing apoptosis.
Figure 6 WZ35 inhibits growth of gefitinib/erlotinib-resistant lung cancer growth in vivo. Nude mice bearing H1975 xenografts received 2.5 or 5 mg/kg WZ35 for 24 days. (A–C) WZ35 treatment inhibits H1975 tumor growth as indicated by tumor volume (A), extracted tumor size (B), and tumor weight (C) (***P<0.001 compared to vehicle control; n=6). (D) Body weight changes of mice in the various experimental groups. (E) Western blot analysis on cleaved caspase 3 levels in lysates prepared from tumors. GAPDH was used as protein loading control. (F) H1975 tumor sections were stained with anti-Ki-67 (brown) to detect proliferating cells. Cells were counterstained with hematoxylin.
Discussion
Mutations in EGFR are major drivers of NSCLC.Citation35 Suppression of EGFR activity through the use of EGFR-TKIs in NSCLC patients harboring EGFR mutations has long been a standard therapeutic regimen. Unfortunately, progression of cancer resulting from primary and acquired resistance to EGFR-TKIs is a critical obstacle in the treatment with NSCLC. Acquired resistance to EGFR-TKIs is caused by several mechanisms, including the acquisition of T790M,Citation36,Citation37 mesencymal epithelial transition factor amplification,Citation38 and EGFR C797S mutation.Citation13 Thus, the discovery of new and effective anticancer agents to tackle gefitinib- and erlotinib-resistance NSCLC is urgent. One potential strategy against EGFR-mutant NSCLC is elevating ROS levels, which has been recently employed to produce methionine 790 oxidation and EGFR degradation in H1975 cells.Citation39 We have also shown that ROS-generating agents cause selective killing pancreatic, colorectal, and gastric cancer cells without serious toxicity to normal cells.Citation19–Citation21 In this study, we first showed that WZ35 rapidly increases ROS generation in H1975 cells. This increased level of ROS led to the activation of the ER stress pathway and disruption of mitochondrial membrane integrity. These perturbations were followed by cell cycle arrest and induction of apoptosis in gefitinib-resistant H1975 cells and xenografts.
ROS play a vital role in the initiation and progression of cancer. Cancer cells exhibit high ROS levels compared with their normal counterparts.Citation40,Citation41 Increased ROS levels in cancer cells are believed to contribute to genomic instability and activation of various signaling cascades related to tumorigenesis. Cancer cells are commonly unable to cope with additional oxidative stress and are likely to be more vulnerable to damage by further increasing the generation and accumulation of ROS induced by exogenous agents.Citation42,Citation43 The increased expression and activity levels of caspases in leukemia cells by arsenic trioxide described earlier.Citation17,Citation18 Thus, accumulating overproduced ROS becomes a useful therapeutic strategy to selectively kill cancer cells without serious toxicity to normal cells.Citation44,Citation45 In our study, we discovered that WZ35 suppressed the growth of NSCLC line and induced apoptosis through the generation of ROS. Neutralizing ROS levels in H1975 cells through NAC prevented WZ35-induced apoptosis, suggesting that ROS produced the observed effects.
The link between elevated ROS levels, activation of the ER stress pathway, and apoptotic death has been demonstrated in various cell lines.Citation46 We have previously shown that WZ35 activates the ER stress pathway in gastric, colon, and pancreatic cancer cells.Citation19–Citation21 In these cancer cells, we noted induction of p-eIF2α and CHOP. The effect of ROS-induced elevations of p-eIF2α and CHOP was reproduced in the gefitinib-resistant H1975. These findings bring forward the possibility to target the UPR pathway for NSCLC patients with acquired resistance to EGFRs. The protein abundances of p-eIF2α, ATF4, and CHOP were elevated 6 h following exposure to WZ35. Associated with the induction of these ER stress-related proteins, distension of the ER can be seen through electron microscopy. CHOP, a member of the growth arrest-and DNA damage-inducible gene (GADD) family, is intricately linked to apoptotic cell death downstream of ER stress. To confirm that CHOP mediates cell apoptosis in H1975, we silenced CHOP through siRNA and then exposed the cells to WZ35. Our results clearly show that WZ35-induced cell death in H1975 is significantly dampened upon producing CHOP deficit. We further showed that WZ35-induced ER stress is abolished when ROS is neutralized by NAC. These findings suggest that ROS production by WZ35 is a critical upstream regulator of ER stress pathway activation, CHOP induction, and cell apoptosis.
In addition to ER stress, changes to mitochondrial membrane potential and permeabilization plays an important role in programmed cell death. The two pathways are also linked whereby ER stress can cause mitochondrial dysfunction and vice versa. In H1975 cells, this ER–mitochondria interface certainly appears to be involved in cytotoxicity of WZ35. We have also demonstrated that the disturbance to mitochondrial membrane integrity induced by WZ35 do share a common upstream signal with the activation of ER stress pathway – ROS. The loss of mitochondrial membrane potential, indicated by the red-to-green shift of JC-1 fluorescence due to changes in dye aggregate-monomer distribution, is followed by the release of cytochrome c leading to the activation of the apoptotic machinery. The exact temporal relationship between ER stress and mitochondrial deficits in H1975 as well as dependencies of these two stress pathways in causing cell death needs to be explored further.
The activation of ROS-mediated apoptosis in cancer cells highlights a new therapeutic strategy for the treatment of gefitinib- and erlotinib-resistant NSCLC. The rationale of this strategy is to elevate ROS to highly toxic levels intracellularly, thereby, activating ROS-induced cell death pathway. In concordant with our cell culture experiments, we have found that WZ35 is also effective in suppressing H1975 tumor growth in a xenograft mouse model. These studies reveal an important role of ROS in the antitumor effects of WZ35 in human NSCLC. Although the direct molecular targets of WZ35 need to be uncovered, our data do suggest that WZ35 may be a promising compound for NSCLC treatment.
Acknowledgments
The work was supported by National Natural Science Foundation of China (81672305 and 81622043), Zhejiang Province Natural Science Funding of China (LY17H160055, LY17H300004, and LYI6H160050), and Wenzhou Science and Technology Project (Y20170176).
Supplementary materials
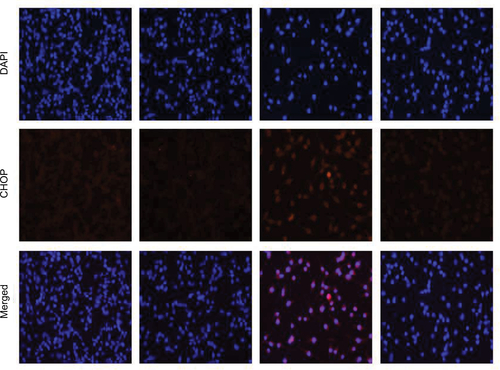
Figure S1 Fluorescence images of H1975 cell lines stained with CHOP. Cells were pretreated with 5 mM NAC for 2 h before exposure to 5 μM WZ35 for 12 h. Cells were stained with DAPI (blue) and CHOP (red).
Abbreviations: NAC, N-acetylcysteine; CHOP, C/EBP-homologous protein; DAPI, 4′,6-diamidino-2-phenylindole.
Disclosure
The authors report no conflicts of interest in this work.
References
- JemalABrayFCenterMMFerlayJWardEFormanDGlobal cancer statisticsCA Cancer J Clin2011612699021296855
- SiegelRLMillerKDJemalACancer statistics, 2017CA Cancer J Clin201767173028055103
- SakashitaSSakashitaMTsaoMSGenes and pathology of non-small cell lung carcinomaSemin Oncol2014411283924565579
- da Cunha SantosGShepherdFATsaoMSEGFR mutations and lung cancerAnnu Rev Pathol20116496920887192
- ZhouCWuYLChenGErlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 studyLancet Oncol201112873574221783417
- RosellRCarcerenyEGervaisRErlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trialLancet Oncol201213323924622285168
- MitsudomiTMoritaSYatabeYGefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trialLancet Oncol201011212112820022809
- LynchTJBellDWSordellaRActivating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinibN Engl J Med2004350212129213915118073
- PaezJGJannePALeeJCEGFR mutations in lung cancer: correlation with clinical response to gefitinib therapyScience200430456761497150015118125
- PaoWMillerVZakowskiMEGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinibProc Natl Acad Sci U S A200410136133061331115329413
- PaoWMillerVAPolitiKAAcquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domainPLoS Med200523e7315737014
- BalakMNGongYRielyGJNovel D761Y and common secondary T790M mutations in epidermal growth factor receptor-mutant lung adenocarcinomas with acquired resistance to kinase inhibitorsClin Cancer Res200612216494650117085664
- YuHATianSKDrilonAEAcquired resistance of EGFR-mutant lung cancer to a T790M-specific EGFR inhibitor: emergence of a third mutation (C797S) in the EGFR tyrosine kinase domainJAMA Oncol20151798298426181354
- SongHNJungKSYooKHAcquired C797S mutation upon treatment with a T790M-specific third-generation EGFR inhibitor (HM61713) in non-small cell lung cancerJ Thorac Oncol2016114e45e4726749488
- HilemanEOLiuJAlbitarMKeatingMJHuangPIntrinsic oxidative stress in cancer cells: a biochemical basis for therapeutic selectivityCancer Chemother Pharmacol200453320921914610616
- ToyokuniSOkamotoKYodoiJHiaiHPersistent oxidative stress in cancerFEBS Lett19953581137821417
- SoignetSLMaslakPWangZGComplete remission after treatment of acute promyelocytic leukemia with arsenic trioxideN Engl J Med199833919134113489801394
- NiuCYanHYuTStudies on treatment of acute promyelocytic leukemia with arsenic trioxide: remission induction, follow-up, and molecular monitoring in 11 newly diagnosed and 47 relapsed acute promyelocytic leukemia patientsBlood199994103315332410552940
- ZhangJFengZWangCCurcumin derivative WZ35 efficiently suppresses colon cancer progression through inducing ROS production and ER stress-dependent apoptosisAm J Cancer Res20177227528828337376
- ZhangXChenMZouPCurcumin analog WZ35 induced cell death via ROS-dependent ER stress and G2/M cell cycle arrest in human prostate cancer cellsBMC Cancer20151586626546056
- ZouPZhangJXiaYROS generation mediates the anti-cancer effects of WZ35 via activating JNK and ER stress apoptotic pathways in gastric cancerOncotarget2015685860587625714022
- ChenWZouPZhaoZSelective killing of gastric cancer cells by a small molecule via targeting TrxR1 and ROS-mediated ER stress activationOncotarget2016713165931660926919094
- ZouPChenMJiJAuranofin induces apoptosis by ROS-mediated ER stress and mitochondrial dysfunction and displayed synergistic lethality with piperlongumine in gastric cancerOncotarget2015634365053652126431378
- ShiYVattemKMSoodRIdentification and characterization of pancreatic eukaryotic initiation factor 2 alpha-subunit kinase, PEK, involved in translational controlMol Cell Biol19981812749975099819435
- LeeESYoonCHKimYSBaeYSThe double-strand RNA-dependent protein kinase PKR plays a significant role in a sustained ER stress-induced apoptosisFEBS Lett2007581224325433217716668
- WalterPRonDThe unfolded protein response: from stress pathway to homeostatic regulationScience201133460591081108622116877
- TabasIRonDIntegrating the mechanisms of apoptosis induced by endoplasmic reticulum stressNat Cell Biol201113318419021364565
- MatsumotoMMinamiMTakedaKSakaoYAkiraSEctopic expression of CHOP (GADD153) induces apoptosis in M1 myeloblastic leukemia cellsFEBS Lett19963952–31431478898082
- GotohTOyadomariSMoriKMoriMNitric oxide-induced apoptosis in RAW 264.7 macrophages is mediated by endoplasmic reticulum stress pathway involving ATF6 and CHOPJ Biol Chem200227714123431235011805088
- MaytinEVUbedaMLinJCHabenerJFStress-inducible transcription factor CHOP/gadd153 induces apoptosis in mammalian cells via p38 kinase-dependent and -independent mechanismsExp Cell Res2001267219320411426938
- ParkSHBlackstoneCFurther assembly required: construction and dynamics of the endoplasmic reticulum networkEMBO Rep201011751552120559323
- PennoAHackenbroichGThieleCPhospholipids and lipid dropletsBiochim Biophys Acta20131831358959423246574
- PintonPGiorgiCSivieroRZecchiniERizzutoRCalcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosisOncogene200827506407641818955969
- WangQWangHJiaYPanHDingHLuteolin induces apoptosis by ROS/ER stress and mitochondrial dysfunction in gliomablastomaCancer Chemother Pharmacol20177951031104128393257
- FabrizioGDi PaolaSStillaAARTC1-mediated ADP-ribosylation of GRP78/BiP: a new player in endoplasmic-reticulum stress responsesCell Mol Life Sci20157261209122525292337
- CampERSummyJBauerTWLiuWGallickGEEllisLMMolecular mechanisms of resistance to therapies targeting the epidermal growth factor receptorClin Cancer Res200511139740515671571
- KobayashiSBoggonTJDayaramTEGFR mutation and resistance of non-small-cell lung cancer to gefitinibN Engl J Med2005352878679215728811
- EngelmanJAZejnullahuKMitsudomiTMET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signalingScience200731658271039104317463250
- LeungELFanXXWongMPTargeting tyrosine kinase inhibitor-resistant non-small cell lung cancer by inducing epidermal growth factor receptor degradation via methionine 790 oxidationAntioxid Redox Signal201624526327926528827
- AcharyaADasIChandhokDSahaTRedox regulation in cancer: a double-edged sword with therapeutic potentialOxid Med Cell Longev201031233420716925
- CairnsRAHarrisISMakTWRegulation of cancer cell metabolismNat Rev Cancer2011112859521258394
- TrachoothamDAlexandreJHuangPTargeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach?Nat Rev Drug Discov20098757959119478820
- NogueiraVHayNMolecular pathways: reactive oxygen species homeostasis in cancer cells and implications for cancer therapyClin Cancer Res201319164309431423719265
- RajLIdeTGurkarAUSelective killing of cancer cells by a small molecule targeting the stress response to ROSNature2011475735523123421753854
- SamoylenkoAHossainJAMennerichDKellokumpuSHiltunenJKKietzmannTNutritional countermeasures targeting reactive oxygen species in cancer: from mechanisms to biomarkers and clinical evidenceAntioxid Redox Signal201319172157219623458328
- SharmaVAndersonDDhawanAZinc oxide nanoparticles induce oxidative DNA damage and ROS-triggered mitochondria mediated apoptosis in human liver cells (HepG2)Apoptosis201217885287022395444