
Abstract
Severe, life-threatening adverse reactions to capecitabine sometimes occur in the treatment of solid tumors. Screening for dihydropyrimidine dehydrogenase (DPYD) deficiency is encouraged before start of treatment, but the genetic variants that are commonly analyzed often fail to explain toxicities seen in clinical practice. Here we describe the case of a 79-year-old Caucasian female with breast cancer who presented with life-threatening, rapidly increasing toxicity after 1 week of treatment with capecitabine and for whom routine genetic DPYD test resulted negative. DPYD exon sequencing found variant c.2242+1G>T at the donor splicing site of exon 19. This variant is responsible for skipping of exon 19 and subsequent generation of a non-functional DPYD enzyme. This variant has not been described previously but was found in three other members of the patient’s family. With this case, we show that exon sequencing of DPYD in patients who experience marked toxicity to fluoropyrimidines and test negative for commonly evaluated variants can prove extremely useful for identifying new genetic variants and better explain adverse reactions causality.
Introduction
Capecitabine is an oral prodrug of 5-fluorouracil (5-FU) that is rapidly and extensively absorbed in the intestine before conversion to 5-FU.Citation1 Severe, life-threatening, and even fatal toxicity has been associated with deficient dihydropyrimidine dehydrogenase (DPYD) activity in patients receiving fluoropyrimidine-based chemotherapy at standard doses.Citation2 Capecitabine-induced adverse reactions include severe hand-foot syndrome, gastrointestinal toxicity, hematological toxicity, and cardiotoxicity.Citation3,Citation4
Low DPYD activity is partly explained by genetic variability in the DPYD gene, which is highly polymorphic.Citation5,Citation6 However, only a few variants (ie, rs3918290 [DPYD*2A], rs55886062 [DPYD*13], and rs67376798) have been clearly associated with severe toxicity to fluoropyrimidines. In our hospital, genotyping of these three variants is available on request for patients starting fluoropyrimidine therapy and has proved to be cost-effective.Citation7 Nonetheless, this test enables us to avoid only 10%–15% of capecitabine-related severe adverse reactions, thus strongly suggesting that other genetic variants in DPYD or in other genes might also be implicated.Citation8 DPYD phenotyping based on the measurement of the enzyme activity in peripheral blood mononuclear cells is another proposed approach to minimize life-threatening toxicity.Citation9 It is yet to be discerned which of these approaches offers the better predictive value and is the most cost-effective to implement or whether a combination of both would be the preferred choice.
Case report
A 79-year-old Caucasian woman was diagnosed with poorly differentiated carcinoma that appeared to have originated in breast tissue. She underwent a second-line chemotherapy based on capecitabine (2 weeks on and 1 week off, total dose of 2,600 mg/day in days 1–14).
After 8 days of treatment, she visited the emergency room with progressive asthenia, grade 1 diarrhea, and grade 1 mucositis and was discharged after parenteral fluid support and intensive treatment with loperamide. Capecitabine was discontinued after 48 hours (cumulative dose 26 g) owing to rapidly increasing toxicity despite oral rehydration and a standard dose of loperamide. The main symptoms were diarrhea (grade 2), nausea (grade 2), odynophagia (grade 2), severe solid dysphagia, painful mucositis (grade 3), and progressive asthenia (grade 2) associated with low-grade fever. She was admitted to hospital immediately for close monitoring with intensive fluid and nutritional support, broad-spectrum antibiotics (piperacillintazobactam, vancomycin, and fluconazole), and pain relievers.
Both hematological toxicity (grade 4 neutropenia and grade 2 thrombocytopenia) and non-hematological toxicity (grade 1 palmar-plantar erythrodysesthesia, grade 2 diarrhea, and grade 3 mucositis) worsened rapidly. Her white blood cell count dropped, reaching a zero nadir at day +16 and remaining unchanged for over 6 days despite treatment with granulocyte colony-stimulating factor. The patient also developed anemia requiring erythrocyte and platelet transfusions. Her platelet nadir started to recover slowly at day +17.
Besides support with blood derivatives, she required third-step analgesia with strong opioids (morphine infusion pump) and intensive nutritional support with parenteral nutrition. The consequences of the severe mucositis and diarrhea resulted in low blood levels of potassium, calcium, phosphate, magnesium, and protein, with significant weight loss (up to 5 kg). In addition, the international normalized ratio was increased, with normal liver transaminases. Hand-foot syndrome progressed more slowly during hospitalization, with progressive, painful swelling and numbness on the palms and soles (up to grade 2), together with progressive alopecia that started 20 days after the withdrawal of capecitabine and lasted for more than 3 months. After 23 days in hospital, the patient was discharged with oral nutritional supplements that were necessary for over 2 months until she could tolerate a normal diet.
DPYD deficiency was suspected as a potential explanation for the life-threatening toxicity that developed after such a short interval of exposure to fluoropyrimidine, in the absence of liver or renal dysfunction and other potential drug interactions. The patient did not receive treatment with fluoropyrimidines again. After two further chemotherapy lines, first cyclophosphamide+ zolendronic acid and later vinorrelbine, the patient died due to disease progression.
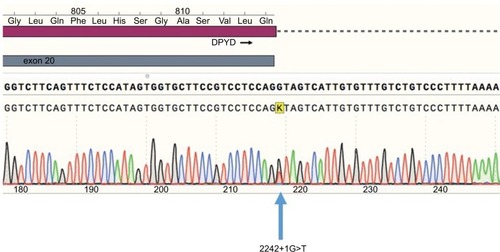
A routine genetic DPYD test was carried out in our hospital because of the strong suspicion of DPYD deficiency. The three DPYD single-nucleotide polymorphisms (SNPs) were genotyped according to the recommendations of the Clinical Pharmacogenetics Implementation Consortium at the time.Citation10 The SNPs were genotyped using real-time PCR and TaqMan probes. None of the three variants were detected. However, the presence of a DPYD deficit continued to be suspected. Therefore, after receiving written informed consent, we performed complete sequencing of the 23 DPYD exons. A total of 23 pairs of oligonucleotides () were used to amplify and sequence DPYD exons. We identified two variants in DPYD exons, c.1601G>A (rs1801158, p.Ser534Asn, DPYD*4) and a previously unreported splice site mutation at position 2242+1G>T, after the end of exon 19 (). This variant was most probably affecting splicing as confirmed using Human Splicing Finder v3.1.Citation11.
Table 1 List of primers used for DPYD exon sequencing
Figure 1 Sequence electropherogram of the 2242+1G>T variant. Blue, dCTP; green, dATP; black, dGTP; red, dTTP.
Abbreviations: dATP, deoxyadenosine triphosphate; dCTP, deoxycytidine triphosphate; dGTP, deoxyguanosine triphosphate; DPYD, dihydropyrimidine dehydrogenase; dTTP, deoxythymidine triphosphate.
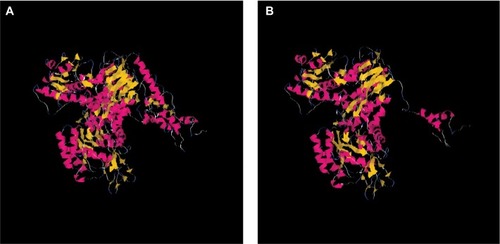
The effect of this second SNP on the donor splicing site was analyzed. RNA was isolated from peripheral blood mononuclear cells and processed to cDNA. A cDNA fragment from exon 18–20 was amplified using specific primers (forward [5′-TTAAAATCTGATGGCACACCTTG] and reverse [5′-TGCTTTTCAGATAAAGCAGGGCT]). PCR product sizes were estimated using electrophoresis. Sequencing of PCR products revealed skipping of exon 19 of DPYD in the patient’s mRNA (). DPYD generated a truncated protein of 795 amino acids lacking the correct sequence from 767 to the end (1025) (). This region is considered essential for the functionality of the protein because it is part of the pyrimidine-binding domain (from 525 to 847).Citation12.
Figure 2 Exon skipping sequence.
Notes: Left: Electrophoresis of amplified cDNA from peripheral blood mononuclear cells of a patient carrying the 2242+1G>T variant. Right, upper: Graphic representation of skipping of exon 19. Right, lower: Sequence electropherogram using the primer Ex18F. A double sequence is observed for 85 nucleotides corresponding to overlapping of exons 19 and 20.
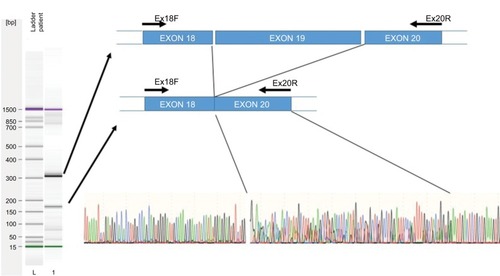
Figure 3 Structures of wild-type DPYD and truncated DPYD.
Notes: The interpretation of mutation effect and the molecular modeling were performed by using Deep View Swiss-PDB viewer and Tasser. (A) Modeling of wild-type DPYD; (B) modeling of truncated DPYD.
Due to the interest of this novel discovery and its potential clinical implications, genotyping of the mutation was later performed in the patient’s offspring: one son, one daughter, and one granddaughter (a daughter descendant). For this purpose, a TaqMan probe was synthesized for 2242+1G>T. All individuals carried the 2242+1G>T mutation in heterozygosis. None of them presented the DPYD* four allele.
DPYD phenotype assessment was performed by pretreatment measurement of plasma uracil (U) and dihydrouracil (UH2) in the descendants.Citation13 Unfortunately, we were not able to analyze a sample from the original patient because at the time this analysis was performed, she was already deceased.
Two samples showed DPYD deficiency according to established criteria (uracil concentration >16 ng/mL and/or UH2/U ratio <6). The other sample showed values that were close to the limit but within the normal activity range. Phenotyping results are shown in .
Table 2 DPYD phenotyping results
A TaqMan probe was synthesized for genotyping of 2242+1G>T in a cohort of 487 anonymous DNA samples from colorectal cancer patients (mainly Caucasians from Iberian ancestry). None of the samples analyzed, except that of the present case, carried this mutation, indicating that it is a rare variant in our setting.
Discussion
The severe toxicity can be explained by the presence of the variant c.2242+1G>T, which generates shorter mRNA and protein, thus rendering a non-functional DPYD protein that lacks a sequence in the pyrimidine-binding domain (). The SNP is just located in the first nucleotide after the end of exon 19. Other DPYD variants affecting donor splicing sites have also demonstrated skipping of the corresponding exonCitation12,Citation14 or generation of an in-frame insertion of small fragments causing severe and occasionally fatal adverse reactions to fluoropyrimidine.Citation15
In this patient, a non-synonymous variant, c.1601G>A (rs1801158, p.Ser534Asn), was also observed. This variant defines the DPYD* four allele and decreases protein activity by 20%. However, although this allele has previously been related to the risk of fluoropyrimidine-induced toxicity,Citation16,Citation17 a recent meta-analysis did not find any significant association between c.1601G>A and severe fluoropyrimidine-induced toxicity.Citation18 The DPYD deficiency observed in two out of three members of the family carrying the c.2242+1G>T mutation supports the effect of this variant on the fluoropyrimidine metabolism. The fact that none of them presented variant c.1601G>A further backs the theory that this new variant may be responsible for the descent in activity. As previously described, individuals carrying one functional and one nonfunctional allele of DPYD are likely to display a partial deficiency with 1.5<UH2/U<6Citation9. The third family member carrying this mutation showed a UH2/U ratio within the normal activity range, although close to the limit (6.6). This could be explained by factors not studied, such as copy number variants increasing the activity of the functional allele, or additional mutations in the other members.
The frequency of the DPYD variant c.2242+1G>T variant is lower than 0.002% in our setting, which suggests that it is a rare variant and its inclusion in our routine genotyping tests would not significantly increase their predictive value. Testing for this variant in other populations (since our sample is mainly integrated by Caucasian individuals of Iberian ancestry) would be very interesting as it could help to investigate its possible origins. This is definitely a line of investigation to be pursued in future works.
Conclusion
We consider the newly described variant 2242+1G>T in DPYD to be the most probable cause of very severe toxicity following the first cycle of treatment with capecitabine in a patient with breast cancer. DPYD phenotyping in patient’s offspring carrying the mutation showed decreased enzymatic activity, further supporting this conclusion. Whole DPYD sequencing is an interesting approach for the identification of causal variants in patients with suspected DPYD deficiency in whom routine DPYD tests yield negative results.
Ethics
Written informed consent was obtained from the three patient’s family members that were tested for DPYD defiency and from all patients in the colorectal cancer cohort that were tested for the presence of c.2242+1G>T. Written informed consent from the patient’s next of kin was provided to have this case published since the patient was deceased at that time.
Acknowledgments
We would like to thank Luis Fernández Pacios at Universidad Politécnica de Madrid, for his support in modeling the DPYD 3D structure. The study was partially supported by the Ministry of Economy and Competitiveness grant PTA2013-8539-I (MIG), ISCIII-FIS grant PI12/00056, “Programa Miguel Servet II grant MSII13/00008” (LALF), and by “Consejería de Educación y Deporte de la Comunidad de Madrid” grant PEJ15/BIO/TL-0603 (CB) and PEJ16/MED/AI-1260 (LALF). All grants were cofunded by the European Regional Development Fund (ERDF [Fondo Europeo de Desarrollo Regional]) Funds from the European Commission “A way of making Europe.” XGG was supported by a grant from Gregorio Marañón Health Research Institute.
Disclosure
The authors report no conflicts of interest in this work.
References
- MercierCCiccoliniJProfiling dihydropyrimidine dehydrogenase deficiency in patients with cancer undergoing 5-fluorouracil/capecitabine therapyClin Colorectal Cancer20066428829617241513
- SaifMWSyrigosKMehraRMattisonLKDiasioRBDeficiency dehydrogenase (DPD) in GI malignancies: experience of 4-yearsPakistan J Med Sci2007236832839
- LeicherLWde GraafJCCoersWTascilarMde GrootJWTolerability of capecitabine monotherapy in metastatic colorectal cancer: a real-world studyDrugs R D201717111712427848234
- SaifMWKatirtzoglouNASyrigosKNCapecitabine: an overview of the side effects and their managementAnticancer Drugs200819544746418418212
- RosmarinDPallesCPagnamentaAA candidate gene study of capecitabine-related toxicity in colorectal cancer identifies new toxicity variants at DPYD and a putative role for ENOSF1 rather than TYMSGut201564111112024647007
- MilanoGHighlight on DPYD gene polymorphisms and treatment by capecitabineScand J Clin Lab Invest Suppl2016245S30S3327454530
- CortejosoLGarcía-GonzálezXGarcíaMIGarcía-AlfonsoPSanjurjoMLópez-FernándezLACost-effectiveness of screening for DPYD polymorphisms to prevent neutropenia in cancer patients treated with fluoropyrimidinesPharmacogenomics201617997998427248859
- TerrazzinoSCargninSDel ReMDanesiRCanonicoPLGenazzaniAADPYD IVS14+1G>A and 2846A>T genotyping for the prediction of severe fluoropyrimidine-related toxicity: a meta-analysisPharmacogenomics201314111255127223930673
- ThomasFHennebelleIDelmasCGenotyping of a family with a novel deleterious DPYD mutation supports the pretherapeutic screening of DPD deficiency with dihydrouracil/uracil ratioClin Pharmacol Ther201699223524226265035
- CaudleKEThornCFKleinTEClinical Pharmacogenetics Implementation Consortium guidelines for dihydropyrimidine dehydrogenase genotype and fluoropyrimidine dosingClin Pharmacol Ther201394664064523988873
- DesmetFOHamrounDLalandeMCollod-BéroudGClaustresMBéroudCHuman Splicing Finder: an online bioinformatics tool to predict splicing signalsNucleic Acids Res2009379e6719339519
- van KuilenburgABMeijerJMaurerDSevere fluoropyrimidine toxicity due to novel and rare DPYD missense mutations, deletion and genomic amplification affecting DPD activity and mRNA splicingBiochim Biophys Acta201718633721730
- Etienne-GrimaldiMCBoyerJCBeroudCNew advances in DPYD genotype and risk of severe toxicity under capecitabinePLoS One2017125e017599828481884
- van KuilenburgABHaasjesJRichelDJClinical implications of dihydropyrimidine dehydrogenase (DPD) deficiency in patients with severe 5-fluorouracil-associated toxicity: identification of new mutations in the DPD geneClin Cancer Res20006124705471211156223
- FlegerMWillomitzerJMeinsmaRDihydropyrimidine dehydrogenase deficiency: metabolic disease or biochemical phenotype?JIMD Rep201737495428275972
- LoganayagamAArenas HernandezMCorriganAPharmacogenetic variants in the DPYD, TYMS, CDA and MTHFR genes are clinically significant predictors of fluoropyrimidine toxicityBr J Cancer2013108122505251523736036
- KleiblZFidlerovaJKleiblovaPInfluence of dihydropyrimidine dehydrogenase gene (DPYD) coding sequence variants on the development of fluoropyrimidine-related toxicity in patients with high-grade toxicity and patients with excellent tolerance of fluoropyrimidine-based chemotherapyNeoplasma200956430331619473056
- MeulendijksDHenricksLMSonkeGSClinical relevance of DPYD variants c.1679T>G, c.1236G>A/HapB3, and c.1601G>A as predictors of severe fluoropyrimidine-associated toxicity: a systematic review and meta-analysis of individual patient dataLancet Oncol201516161639165026603945