
Abstract
Purpose
Multiple gastrointestinal stromal tumors (GISTs) are rare. The aim of this study was to investigate the clinicopathologic characteristics and prognosis of multiple GISTs.
Patients and methods
Between May 2003 and June 2018, patients who underwent surgery for multiple GISTs were retrospectively analyzed. Exons 9, 11, 13, and 17 of the KIT gene, and exons 12, and 18 of the PDGFRA gene were examined in 34 tumors from 20 patients.
Results
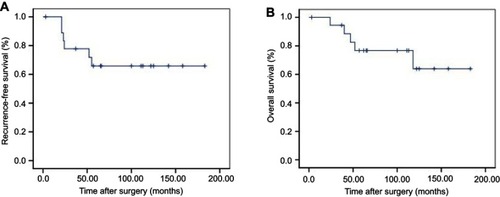
A total of 20 patients with multiple GISTs were enrolled. There were 11 females and nine males with a median age of 59 years (range: 37–80 years). Of these cases, 16 were sporadic cases and four were associated with GIST syndromes (two cases of Carney triad and two cases of neurofibromatosis type 1 [NF1]). The most common presentation was gastrointestinal bleeding. Carney triad GISTs did not exhibit KIT/PDGFRA mutations. One of the NF1 patients was a KIT/PDGFRA wild-type, and the other patient had a PDGFRA mutation. Of the sporadic cases, one shared the same KIT gene mutation within each GIST and one had two lesions that were both wild-type for KIT and PDGFRA. Different KIT mutations among individual tumors were detected in seven patients. During the median follow-up period of 66 months (range: 3–183 months), four patients developed liver or abdominal metastases, three of whom expired due to the disease. The rates of recurrence-free survival and overall surviva at 5 years were 65.8% and 76.7%, respectively.
Conclusion
Multiple GISTs may occur as sporadic tumors or as an additional component of specific syndromes (eg, Carney triad and NF1) that display different clinicopathologic characteristics based on their particular underlying mechanisms. The overall prognosis of patients with multiple GISTs is comparable to that of patients with only a single GIST.
Introduction
Gastrointestinal stromal tumors (GISTs) are rare neoplasms that originate from the interstitial cells of Cajal in the gut, with an annual incidence of 10–15 per million.Citation1,Citation2 While GISTs can occur in any part of the gut, they are most common in the stomach and small intestine, and less frequent in the colorectum and esophagus.Citation2 Some rare tumors may arise in the mesentery and omentum, which are referred to as extra-GISTs (EGISTs).Citation3 The majority of GISTs have an activating mutation in either KIT or platelet-derived growth factor A (PDGFRA).Citation1,Citation4 GISTs are solitary tumors that commonly occur in individuals between 55 and 65 years of age.Citation2 The occurrence of multiple GISTs is very rare and is usually only observed in specific tumor syndromes such as familial GIST syndrome,Citation5 neurofibromatosis type 1 (NF1),Citation6 and the Carney triad.Citation7 Although their pathogenesis and clinical manifestations are different, these tumor syndromes confer a high risk for developing multiple neoplasms. Multiple sporadic GISTs may also be observed outside of the familial and syndromic settings, and most of them could be easily misinterpreted as recurrent or metastatic disease.Citation8–Citation12 However, data on the clinicopathologic features of multiple GISTs are currently limited. In this study, we performed a clinicopathologic and prognostic analysis of multiple GISTs.
Patients and methods
Patient identification
The clinicopathologic data of patients who underwent resection of multiple GISTs between May 2003 and June 2018 at The First Affiliated Hospital, Zhejiang University School of Medicine (Hangzhou, China), were retrospectively collected. The diagnosis of multiple GISTs was defined as two or more GISTs appearing in one or more different organs without evidence of metastasis or recurrence.Citation8 Genomic DNA was independently extracted from formalin-fixed, paraffin-embedded tumor tissue of individual lesions collected from 20 patients. Amplification using polymerase chain reaction and direct sequencing of KIT exons 9, 11, 13, and 17, as well as PDGFRA exons 12, and 18 were successfully performed in 34/54 GISTs.
Ethical approval
The study was approved by the ethical committee of The First Affiliated Hospital, Zhejiang University School of Medicine, (Hangzhou, China), and the study was conducted in accordance with the tenets of the Declaration of Helsinki. The number of the approval document is 20181035. The data involved in this study was retrospectively collected from hospital electronic system and did not include any personal information or privacy. Therefore, the ethical committee of The First Affiliated Hospital, Zhejiang University School of Medicine determined that the patient consent to review their medical records was not required. Furthermore, we stated that all data from patients was anonymized or maintained with confidentiality.
Statistical analysis
Overall survival (OS) was calculated from the date of operation to the date of death (regardless of the cause). Recurrence-free survival (RFS) was calculated from the date of operation to the date of disease recurrence or death (regardless of the cause). The results are presented as percentages or medians (ranges), and survival curves were generated using the Kaplan-Meier method. All data were analyzed using the SPSS software package (ver.19.0; SPSS Inc., Chicago, IL, USA).
Results
As shown in , 11 females and nine males with a median age of 59 years (range: 37–80 years) were included in this study. These 20 patients represented 1.1% of 1,800 consecutive GIST cases managed in our hospital. Bleeding (melena and hematemesis) and abdominal pain were the most frequent symptoms. One female (case 5) had an asymptomatic palpable abdominal tumor at presentation. Among the 20 patients with multiple GISTs, 16 were sporadic and four were associated with GIST syndromes. The Carney triad was observed in two young females who had multiple gastric GISTs and pulmonary chondromas (cases 1 and 2). In case 2, computed tomography revealed a 2-cm left adrenal adenoma, while a small intestinal GIST was incidentally found intraoperatively. Two females (cases 7 and 11) were associated with NF-1, which was confirmed by skin biopsy of cutaneous neurofibromas; one (case 7) had six tumors in the small intestine, and the other (case 11) had one tumor in the stomach and five in the small intestine.
Table 1 Clinicopathologic findings of patients with multiple GISTs
Overall, 54 tumors were detected in the 20 patients included in this study: 25 (46.3%) in the stomach, 23 (42.6%) in the small intestine, three (5.6%) in the rectum, and three (5.6%) in the mesentery. The number of tumors was two in 12 patients, three in six patients, and six in two patients. Seventeen patients had lesions located in a single organ (10 patients in the stomach, five patients in the small intestine, one patient in the rectum, and one patient in the mesentery) and the other three patients had lesions involving two organs (stomach and small intestine; cases 2, 3, and 7).
The tumor size ranged from 0.3 to 17.0 cm(mean, 3.4 cm), excluding three tumors that were <1 mm (cases 2, 10, and 18). Of these tumors, 30 (55.6%) were ≤2 cm, 14 (25.9%) were >2 cm and ≤5 cm, six (11.1%) were >5 cm and ≤10 cm, and four (7.4%) were >10 cm. The mitotic count per 50 high power fields (HPFs) was <5 in 31 (57.4%) GISTs, 5–10 in 19 (35.2%) GISTs, and >10 in four (7.4%) GISTs. According to the modified National Institutes of Health classification system,Citation13 the risk of recurrence was very low in 19 (35.2%) GISTs, low in eight (14.8%) GISTs, intermediate in 12 (22.2%) GISTs, and high in 15 (27.8%) GISTs. Based on cell morphology, the tumors were classified as spindle (85.2%), mixed (13.0%), or epithelioid (1.9%). Immunohistochemically, 98.1%, 87.0%, and 31.5% of the GISTs were positive for CD117, CD34, and SMA, respectively.
Of the 34 genetically studied GISTs, 11 (32.4%) and 10 (29.4%) GISTs showed somatic mutations in the KIT and PDGFRA genes, respectively (). There were no germline mutations detected in the KIT and PDGFRA genes. Mutational analyses of tumor tissue from two patients (cases 1 and 2) with Carney triad revealed wild-type sequences. We examined KIT and PDGFRA mutations in six lesions from two NF1 patients (cases 7 and 11). One lesion derived from case 7 did not have detectable KIT or PDGFRA mutations, while five lesions derived from case 11 shared the same mutation at exon 18 of PDGFRA (p.V824V). One patient (case 5) had two lesions that were wild-type for both KIT and PDGFRA, without clinicopathological signs of NF1 or Carney triad. Three patients (cases 6, 10, and 18) had both mutation-positive and wild-type lesions. In case 19, the two lesions carried an identical KIT mutation. Different mutations were detected in seven patients, and the mutational status for matched lesions was as follows: KIT exon 11/PDGFRA exon 18 (n=3), KIT exon 11/wild-type (n=2), KIT exon 11/KIT exon 11 (n=1), and PDGFRA/wild-type (n=1). There were four patients (cases 4, 8, 12, and 15) with only one lesion successfully sequenced. The remaining three patients did not have available sequencing data of the KIT or PDGFRA genes.
After a median follow-up period of 66 months (range: 3–183 months), 13 patients were alive without tumor recurrence. Two patients (cases 15 and 19) expired due to the disease with metastasis to the liver at 22 and 118 months after the surgery, respectively. Two patients (cases 6 and 8) had metastases in the peritoneal cavity, one (case 6) was alive with disease after a follow-up of 62 months whereas the other (case 8) expired due to the disease after a follow-up of 40 months. One patient (case 13)underwent emergency surgery for intestinal obstruction caused by GISTs and expired due to severe abdominal infection after surgery. The remaining two patients (cases 14 and 16) expired due to unknown causes. The rates of recurrence-free survival and overall survival at 5 years were 65.8% and 76.7%, respectively ( and ).
Figure 1 Progression-free survival (A) and overall survival (B) for patients with multiple GISTs.
Four patients (cases 1, 2, 7, and 19) received adjuvant therapy with imatinib after the surgery for 2, 2, 1, and 1 years, respectively. Case 19 underwent resection of liver metastases 55 months after resection of the primary tumor. Subsequently, the patient continued to receive imatinib for 2 years and expired due to tumor progression. One patient (case 6) initiated treatment with imatinib after developing intraperitoneal metastases 21 months after surgery. The condition of the patient was reportedly stable at the last follow-up.
Discussion
The majority of GISTs occur as sporadic solitary neoplasms resulting from somatic mutations of KIT or PDGFRA.Citation1,Citation4 In our search for patients with multiple GISTs in 1800 consecutive patients managed in our hospital, we found two patients with NF1, two patients with Carney triad, and 16 patients with sporadic multiple GISTs. Multiple GISTs comprised of 1.1% of GIST cases in our consecutive series. As suggested by previous studies, in cases of familial GIST syndrome,Citation5 NF1,Citation6,Citation8 or the Carney triad,Citation7 patients often develop multiple GISTs with distinct characteristic features. Moreover, tumors in these cases generally develop at an early age and either carry germline KIT or PDGFRA mutations (familial GISTs) or lack such mutations.Citation5–Citation8 Different types of KIT and PDGFRA mutations were observed in seven patients, suggesting that these patients may simply represent the simultaneous occurrence of independent sporadic GISTs.
NF1 is an autosomal dominant disease caused by germline NF1 mutations characterized by neurofibromas and café-au-lait spots.Citation6 The loss of heterozygosity (LOH) at 14q and 22q, together with activation of the Ras-MAPK pathway, may contribute to the development of GIST in NF1.Citation22 Most NF1-associated GISTs present as numerous small asymptomatic tumors with a benign clinical course, mainly in the small intestine. These GISTs rarely carry KIT or PDGFRA mutations.Citation6,Citation8 In a previous study investigating 252 GISTs in 126 patients with NF1, only 8 GISTs (4.8%) showed KIT and PDGFRA mutations.Citation25 In case 11, a mutation at codon 824 coding for valine in exon 18 of PDGFRA was confirmed using sequencing as c. 2472 C>T, p. V824V. In our study, we described two older females with NF1, both of whom had six tumors with a mitotic count <5/50 HPFs and median tumor sizes of 2 and 1 cm, respectively. The characteristics described in the present study were compatible with those of cases presented in previous reports.Citation6,Citation8,Citation25
Contrary to NF1, the Carney triad is a non-heritable syndrome that consists of multiple gastric GISTs, paragangliomas, and pulmonary chondromas.It is mainly observed in young females.Citation7 The mechanism of tumor development includes epigenetic inactivation of succinate dehydrogenase C (SDHC) through hypermethylation.Citation23 Notably, 72% of patients only exhibit two of the three components of the triad, with gastric and pulmonary tumors being the most common combination.Citation7 The aforementioned two young females(cases 1 and 2) had pulmonary chondromas and multifocal gastric GISTs. A recent study described that adrenal neoplasm was a fourth component of the syndrome.Citation16 The present study corroborated this finding (case 2). Similar to a previous study,Citation15 our study also indicated that GISTs in the Carney triad are negative for KIT and PDGFRA mutations. Thus, imatinib may not be an effective treatment option for patients with the Carney triad.Citation14,Citation15 However, Chen et alCitation18 previously reported that a Carney triad patient with metastasis achieved a stable condition during treatment with imatinib for 32 months. In the current study, after 2 years of treatment with imatinib, there was no recurrence during the post-operative follow-up period. Future studies are warranted to elucidate the effects of imatinib therapy for GISTs in the Carney triad.
Multiple sporadic GISTs outside of familial and syndromic settings are rarely described.Citation8–Citation12 In our study, nine males and seven females with a median age of 66 years were diagnosed with multiple sporadic GISTs. They were slightly more prevalent in males than females (1.3:1), in accordance with the findings reported by Agaimy et al.Citation12 In most cases, the size of synchronous lesions from the same patient differed greatly, in contrast to the findings reported by Gasparotto et al.Citation10 Frequently, large masses produce symptoms such as abdominal pain and bleeding, while small masses are asymptomatic and incidentally detected during surgery. Notably, the largest lesion in our study was detected in the mesentery, mainly because small GISTs rarely result in clinical manifestations.Citation3 Small GISTs with a diameter <1 cm are defined as micro-GISTs.Citation17 The features of micro-lesions reported in our patients included location in the stomach, few mitoses, and benign histology. These findings were in accordance with the results of a study conducted by Kawanoma et al.Citation19 In our study, the lesions were detected in one or more organs in each patient, as previously reported.Citation10–Citation12 Multiple sporadic GISTs had a predilection for the stomach (54.1%) over the small intestine (29.7%). Similar to solitary neoplasms, multiple sporadic GISTs have a varying potential for malignancy, ranging from virtually indolent tumors to fatal cancers. It was reported that the estimated 5-year RFS rate for GISTs was 70.5%.Citation24 In our study, the 5-year RFS rate for multiple GISTs was 66%, which is comparable to that of patients with only a single tumor.
Currently, there are no established criteria for confirming the diagnosis of primary versus metastatic tumors in patients initially presenting with multiple tumors. Kang et alCitation8 were the first to use KIT or PDGFRA genes as markers of tumor clonality, and described one patient with two jejunal GISTs harboring different types of KIT mutations. Similarly, Haller et alCitation9 analyzed four patients with one clinically apparent tumor and 1–3 additional small incidental tumors. Different patterns of KIT or PDGFRA mutation were found in various paired lesions, further indicating that the occurrence of multiple sporadic GISTs may represent a synchronous occurrence of independent sporadic GISTs. Moreover, microsatellite analysis of tumors with different KIT mutation types revealed that distinct mutations in multiple GISTs indicate independent primary tumors.Citation8–Citation10 In the current study, different patterns of KIT or PDGFRA mutation were observed in seven patients, indicating an independent origin of these tumors. Therefore, current studies tend to support the field carcinogenesis theory, which hypothesizes that multiple sporadic GISTs originate from distinct subsets of interstitial cells of Cajal that carry KIT/PDGFRA mutations.Citation10,Citation12 However, for independent GISTs harboring the same KIT or PDGFRA mutations or lacking mutations, we were unable to assess the clonal relationship between the lesions through mutation analysis.Citation10,Citation12 Agaimy et alCitation11 analyzed 14 GISTs from seven patients, and reported that the involvement of the muscularis propria was a potentially reliable marker for primary GISTs. Therefore, molecular analysis in combination with histological features may assist in establishing a correct diagnosis.
Radical surgery remains the mainstay of treatment for multiple GISTs, while the administration of imatinib is recommended for the treatment of metastatic GISTs.Citation17 In surgery, it is important to evaluate the number and sites of tumors for the removal of all lesions. When considering the use of imatinib, molecular analysis is critical because it assists in excluding insensitive mutation types (eg PDGFRA D842V mutation) and allows the use of higher doses for KIT exon 9 mutations.Citation20,Citation21 Given that multiple GISTs may exhibit variable sensitivity to drugs due to the presence of different mutations, mutation analysis of different lesions is necessary.
Conclusion
Multiple GISTs may occur as sporadic tumors or as an additional component of specific syndromes, such as the Carney triad and NF1. These GISTs exhibit different clinicopathologic characteristics based on their underlying mechanisms. The prognosis of patients with multiple GISTs is similar to that of patients with solitary tumors.
Disclosure
The authors declare that they have no conflicts of interest in this work.
Acknowledgment
The authors appreciate Dr Zhang Y, Department of Urology, The First Affiliated Hospital, Zhejiang University School of Medicine, for his assistance in writing the manuscript. The manuscript was edited for correct English language usage, grammar, punctuation and spelling by qualified native English speaking editors at The Charlesworth Group.
References
- Corless CL, Barnett CM, Heinrich MC. Gastrointestinal stromal tumours: origin and molecular oncology. Nat Rev Cancer. 2011;11(12):865–878. doi:10.1038/nrc314322089421
- Søreide K, Sandvik OM, Søreide JA, et al. Global epidemiology of gastrointestinal stromal tumours (GIST): a systematic review of population-based cohort studies. Cancer Epidemiol. 2016;40:39–46. doi:10.1016/j.canep.2015.10.03126618334
- Kim KH, Nelson SD, Kim DH, et al. Diagnostic relevance of overexpressions of PKC-θ and DOG-1 and KIT/PDGFRA gene mutations in extragastrointestinal stromal tumors: a Korean six-centers study of 28 cases. Anticancer Res. 2012;32(3):923–937.22399613
- Heinrich MC, Corless CL, Duensing A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299(5607):708–710. doi:10.1126/science.107966612522257
- Neuhann TM, Mansmann V, Merkelbach-Bruse S, et al. A novel germline KIT mutation (p.L576P) in a family presenting with juvenile onset of multiple gastrointestinal stromal tumors, skin hyperpigmentations, and esophageal stenosis. Am J Surg Pathol. 2013;37(6):898–905. doi:10.1097/PAS.0b013e31827bc07123598963
- Agaimy A, Vassos N, Croner RS. Gastrointestinal manifestations of neurofibromatosis type 1 (Recklinghausen’s disease): clinicopathological spectrum with pathogenetic considerations. Int J Clin Exp Pathol. 2012;5(9):852–862.23119102
- Carney JA. Gastric stromal sarcoma, pulmonary chondroma, and extra-adrenal paraganglioma (Carney triad): natural history, adrenocortical component, and possible familial occurrence. Mayo Clin Proc. 1999;74(6):543–552. doi:10.4065/74.6.54310377927
- Kang DY, Park CK, Choi JS, et al. Multiple gastrointestinal stromal tumors: clinicopathologic and genetic analysis of 12 patients. Am J Surg Pathol. 2007;31(2):224–232. doi:10.1097/01.pas.0000213318.66800.9417255767
- Haller F, Schulten HJ, Armbrust T, et al. Multicentric sporadic gastrointestinal stromal tumors (GISTs) of the stomach with distinct clonal origin: differential diagnosis to familial and syndromal GIST variants and peritoneal metastasis. Am J Surg Pathol. 2007;31(6):933–937. doi:10.1097/01.pas.0000213440.78407.2717527083
- Gasparotto D, Rossi S, Bearzi I, et al. Multiple primary sporadic gastrointestinal stromal tumors in the adult: an underestimated entity. Clin Cancer Res. 2008;14(18):5714–5721. doi:10.1158/1078-0432.CCR-08-0622
- Agaimy A, Märkl B, Arnholdt H, et al. Multiple sporadic gastrointestinal stromal tumours arising at different gastrointestinal sites: pattern of involvement of the muscularis propria as a clue to independent primary GISTs. Virchows Arch. 2009;455(2):101–108. doi:10.1007/s00428-009-0803-119572146
- Agaimy A, Dirnhofer S, Wünsch PH, et al. Multiple sporadic gastrointestinal stromal tumors (GISTs) of the proximal stomach are caused by different somatic KIT mutations suggesting a field effect. Am J Surg Pathol. 2008;32(10):1553–1559. doi:10.1097/PAS.0b013e31817587ea18724245
- Joensuu H. Risk stratification of patients diagnosed with gastrointestinal stromal tumor. Hum Pathol. 2008;39(10):1411–1419. doi:10.1016/j.humpath.2008.06.02518774375
- Chen LL, Trent JC, Wu EF, et al. A missense mutation in KIT kinase domain 1 correlates with imatinib resistance in gastrointestinal stromal tumors. Cancer Res. 2004;64(17):5913–5919. doi:10.1158/0008-5472.CAN-04-008515342366
- Matyakhina L, Bei TA, McWhinney SR, et al. Genetics of carney triad: recurrent losses at chromosome 1 but lack of germline mutations in genes associated with paragangliomas and gastrointestinal stromal tumors. J Clin Endocrinol Metab. 2007;92(8):2938–2943. doi:10.1210/jc.2007-079717535989
- Carney JA, Stratakis CA, Young WF. Adrenal cortical adenoma: the fourth component of the Carney triad and an association with subclinical Cushing syndrome. Am J Surg Pathol. 2013;37(8):1140–1149. doi:10.1097/PAS.0b013e318285f6a223681078
- Joensuu H, Hohenberger P, Corless CL. Gastrointestinal stromal tumour. Lancet. 2013;382(9896):973–983. doi:10.1016/S0140-6736(13)60106-323623056
- Chen CF, Chuang CH, Liu MK, et al. Clinical, radiologic and pathologic characteristics of the carney triad: a case report and literature review. Kaohsiung J Med Sci. 2010;26(8):428–434. doi:10.1016/S1607-551X(10)70069-320705254
- Kawanowa K, Sakuma Y, Sakurai S, et al. High incidence of microscopic gastrointestinal stromal tumors in the stomach. Hum Pathol. 2006;37(12):1527–1535. doi:10.1016/j.humpath.2006.07.00216996566
- Joensuu H, Eriksson M, Sundby Hall K, et al. One vs. three years of adjuvant imatinib for operable gastrointestinal stromal tumor. JAMA. 2014;307(12):1265–1272. doi:10.1001/jama.2012.347
- Debiec-Rychter M, Sciot R, Le Cesne A, et al. KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur J Cancer. 2006;42(8):1093–1103. doi:10.1016/j.ejca.2006.01.03016624552
- Yamamoto H, Tobo T, Nakamori M, et al. Neurofibromatosis type 1-related gastrointestinal stromal tumors: a special reference to loss of heterozygosity at 14q and 22q. J Cancer Res Clin Oncol. 2009;135(6):791–798. doi:10.1007/s00432-008-0514-z19020900
- Haller F, Moskalev EA, Faucz FR, et al. Aberrant DNA hypermethylation of SDHC: a novel mechanism of tumor development in Carney triad. Endocr Relat Cancer. 2014;21(4):567–577. doi:10.1530/ERC-14-025424859990
- Joensuu H, Vehtari A, Riihimäki J, et al. Risk of recurrence of gastrointestinal stromal tumour after surgery: an analysis of pooled population-based cohorts. Lancet Oncol. 2012;13(3):265–274. doi:10.1016/S1470-2045(12)70140-722153892
- Salvi PF, Lorenzon L, Caterino S, et al. Gastrointestinal stromal tumors associated with neurofibromatosis 1: a single centre experience and systematic review of the literature including 252 cases. Int J Surg Oncol. 2013;2013:398570.24386562