
Abstract
In the last decade, the inhibition of the mechanistic target of Rapamycin (mTOR) in renal clear cell carcinoma (RCC) has disappointed the clinician’s expectations. Many clinical trials highlighted the low efficacy and unmanageable safety profile of first-generation mTOR inhibitors (Rapalogs), thus limiting their use in the clinical practice only to those patients who already failed several therapy lines. In this review, we analyze the major resistance mechanisms that undermine the efficacy of this class of drugs. Moreover, we describe some of the possible strategies to overcome the mechanisms of resistance and their clinical experimentation, with particular focus on novel mTOR inhibitors and the combinations of mTOR inhibitors and other anti-cancer drugs.
Introduction
The treatment of metastatic Renal Cell Carcinoma (mRCC) has improved drastically over the last two decades. In fact, from a scarcity of specific therapeutic options, we assisted to the rise and the affirmation of a number of immune checkpoint inhibitors (ICIs) and small molecule antiangiogenic targeted agents, given either in combination or as a monotherapy, which dramatically changed the natural history of this once orphan disease.
Among all the molecular targets identified and successfully drugged in recent years, however, the mechanistic Target of Rapamycin (mTOR, previously mammalian Target of Rapamycin), has a pretty unique story. mTOR pathway controls several aspects of cell proliferation, migration, and metabolism, to an extent that is still largely unknown. The first identified mTOR inhibitor was, obviously, Rapamycin. This macrolide was first discovered in 1975 in a soil sample from the Easter Island of the South Pacific, also known as Rapa Nui (hence the name).Citation1
In the early ‘90s, the mechanistic action of Rapamycin was clarified, leading to its application as an immunosuppressor and to the first in vitro studies on its anti-cancer properties. Hence, the first generation of Rapamycin derivatives, with reduced immunosuppressive effect, was developed. The two most promising molecules of this class, Temsirolimus and Everolimus (the so-called Rapalogs), have proven effective in several preclinical models. However, in clinical trials, they succeeded only in specific cancers, including mRCC. Moreover, the extent of this success was much less than expected. Rapalogs indeed showed a primarily cytostatic action, suggesting that their role should be disease stabilization rather than tumor regression. This has been attributed to the only partial inhibition of mTOR that fails to block the feedback loop between mTOR and its client kinase AKT, which arises as a mechanism of resistance.
In clinical trials involving mRCC, Rapalogs demonstrated superiority when compared with old therapeutic strategies, such as interferon-alpha. However, across the last decade, Rapalogs were heavily outclassed by multi-kinase inhibitors and immune checkpoint inhibitors. Therefore, they now find an application only after the failure of these drugs. On the other hand, more specific and potent mTOR inhibitors are currently being developed and tested, learning from the failures of the first generation. This review aims to analyze whether is it possible to overcome the resistance to mTOR inhibitors and what strategies should be preferred in the pursuit of this goal.
The Role of mTOR in RCC
mTOR is an intracellular serine/threonine kinase that regulates several aspects of cell growth, proliferation, movement, and survival.Citation2 mTOR is able to react to many diverse stimuli (such as growth factors, cell nutritional status, and cellular stress) producing different responses. Specifically, mTOR exists in two distinct complexes, rapamycin-sensitive mTOR complex 1 (mTORC1) and rapamycin-sensitive mTOR complex 2 (mTORC2), that exert different effects, thus explaining the pleiotropy of this molecule. mTORC1 is formed by mTOR, regulatory-associated protein of mTOR (Raptor), G-protein β-subunit-like protein/LST8 (GbL), and proline-rich AKT substrate 40 kDa (PRAS40); by modulating specific transcriptional factors, mTORC1 modulates lipids, protein and nucleotide biosynthesis, cell growth, autophagy, mitochondrial metabolism, and hypoxic response.Citation3,Citation4
On the other hand, mTORC2 is composed of GbL, the protein rapamycin-insensitive companion of mTOR (Rictor), and mammalian stress-activated protein kinase (SAPK)-interacting protein (mSIN1); it plays a role in metabolic rewiring and cytoskeletal organization, being involved in cell movement.Citation4
These intracellular serine/threonine kinases regulate many important cellular mechanismsCitation5 and the inability to regulate the mTOR pathway is emerging as a common feature in several malignancies. The abnormal activation of the mTOR signaling pathway results in an unbalance in cell growth signals, leading to carcinogenesis. In most cancer cells, uncontrolled mTOR signaling provides a selective growth advantage.
The PIK3CA/AKT/mTOR pathway is one of the most commonly activated pathways in human cancers.Citation5 Mutations that trigger the pathologic activation of mTOR have been found in several types of cancer, albeit this axis is of uttermost importance in RCC,Citation6 a heterogeneous group of cancers originating from the nephron. There are several histotypes of kidney tumors, the most frequent being clear cell (65%), followed by the papillary subtype (20%) and the chromophobe subtype (5%).Citation7 Each histotype harbors its specific mutations, clinical characteristics, and therapeutic responses.Citation8
Many studies aimed at identifying the gene mutations responsible for the development of RCC have been conducted over the years. A major role in clear cell RCC (ccRCC) genesis is played by the loss of function of the von Hippel-Lindau (VHL) gene, a tumor suppressor whose main role consists in inducing the degradation of the Hypoxia Inducible Factor (HIF). The second most studied mechanism involves the mutations affecting the PI3K/AKT/mTOR signaling pathway. These mutations either promote the overactivation of the PI3K/AKT/mTOR signaling cascadeCitation9 or are responsible for the inhibition of the PTEN, a tumor suppressor gene that plays an inhibitory role on the aforementioned axis.Citation10 PTEN, PI3K, AKT, and mTOR represent fundamental checkpoints of the signal transduction pathway involved in the control of cell development and proliferation, being heavily implied in RCC pathogenesis and progression.Citation11,Citation12 In fact, aberrations in the PI3K/AKT/mTOR pathway can be detected in 28% of clear cell RCC cases.Citation13 It’s worth noting that mTOR mutation is particularly prevalent in highly vascularized tumor areas since vascularization allows nutrients and growth factors to activate the mTOR pathway.Citation14–Citation17
Mutations of the mTOR axis are also described in uncommon RCC subtypes, such as chromophobe RCC,Citation18 acquired cystic disease-associated RCC, eosinophilic solid and cystic RCC, RCC with fibromyomatous stroma and even in the low-grade oncocytic tumor.Citation19 Although the idea of a mTOR-driven RCC subclass is appealing, further research is needed in order to understand whether this mutations are to be considered driver or passenger mutations.
Whether these mutations can impact the survival of patients with RCC, is a debated topic. Fan et al demonstrated that the increased expression of mTOR and its related proteins impacts positively on survival.Citation20 On the other hand, Ocana et al concluded that mutations of the mTOR axis, especially those implying the loss of PTEN, correlate with a significantly worse prognosis.Citation21 Regardless of its significance as a prognostic factor, mTOR remains a pivotal regulator of cell growth and proliferation, being thus considered a potential druggable target.
Indeed, in the past decades, different specific mTOR inhibitors were developed.
mTOR Inhibitors in Current Clinical Practice
Since dysregulation of the PI3K/AKT/mTOR signaling pathway is frequently encountered in patients with RCC,Citation22 mTOR inhibitors were largely studied for the treatment of locally advanced or metastatic RCC.Citation17,Citation23 Rapamycin was the first identified mTOR inhibitor. It acts by forming a complex with the protein FKBP12, which acts as the effective inhibitor of mTORC1.Citation24 From preclinic data, it’s known that Rapamycin can slow cell growth and proliferation, or act synergistically with chemotherapy to induce cell death. Moreover, suppressing the mTOR pathway will slow down the consumption of external resources, inducing autophagy. Paradoxically, even an activator of the mTOR pathway could be therapeutically beneficial, because it would push cells to run out of energy and nutrients, and in the absence of stimuli, it could no longer maintain essential life processes.Citation25,Citation26 In addition, Rapamycin was proven to inhibit metastasis of RCC in an in-vivo model.Citation27
Although Rapamycin di per se was never extensively studied, as a therapeutic agent, in mRCC patients, its derivatives were largely used in this disease.
Everolimus
Amato et alCitation28 first carried on a Phase II trial of Everolimus in patients with mccRCC that received only 1 or no previous treatment lines. Everolimus reported an mPFS of 11.2 months, with an objective response rate (ORR) of 14% and a disease control rate (DCR) of 87%; this study represented the proof of principle of Everolimus activity in pre-treated mRCC patients.
In the RECORD-1 Phase III trial, Motzer et alCitation29 randomized mccRCC patients progressing after one or more previous lines of therapy (including at least a previous TKI, mostly Sunitinib) to receive either Everolimus or placebo. Everolimus yielded a significantly longer median PFS (4.9 months vs 1.9 months, HR 0.33, p<0.00001), with an acceptable safety profile. However, it should be noted that in this study, the crossover between arms was allowed, and was massive. As a consequence, no OS benefit was observed.
Everolimus was tested also in naive mccRCC patients. In the RECORD-2 randomized phase II trial,Citation30 previously untreated mccRCC patients were randomized to receive either the combination of Everolimus + Bevacizumab (an anti-VEGFR monoclonal antibody) or Interferon + Bevacizumab. The median PFS was 9.3 vs 10 months (HR = 0.90, 95% CI 0.71–1.15; P = 0.423).
More recently, the RECORD-3 phase II trialCitation31 compared the sequence of Everolimus followed by Sunitinib with the reverse sequence (ie Sunitinib followed by Everolimus) in naïve mRCC patients (including a subpopulation of non-clear cell RCC). Sunitinib upfront resulted in a higher median PFS (10 months vs 8 months) and ORR (27% vs 8%) when compared with Everolimus upfront. In addition, the combined median PFS of the two sequences was higher for that which started with Sunitinib (25.8 months vs 21.1 months). From all these findings it can be inferred that the place for Everolimus should be in the second-line, after the failure of a first-line with TKI. However, more recent trials furtherly scaled back the role of this drug.
In fact, the METEOR phase III trialCitation32 compared Cabozantinib and Everolimus in mccRCC patients progressing after one or more VEGFR tyrosine kinase inhibitors. Cabozantinib reported a much better performance both in terms of mPFS (7.4 months vs 3.9 months) and ORR (17% vs 3%). In the same setting, Everolimus was compared with the immune checkpoint inhibitor Nivolumab.
Furthermore, the Checkmate-025 phase III trialCitation33 enrolled patients that received previous treatment with one or two regimens of antiangiogenic therapy and randomized them to receive either Nivolumab or Everolimus. Although the two drugs performed similarly in terms of median PFS (4.6 months vs 4.4 months), a significant difference was observed in the ORR (25% vs 5%) and median OS (25 months vs 19.6 months, HR = 0.73 (98.5% CI, 0.57 to 0.93; P=0.002), probably due to the development of long-lasting specific immune responses. Therefore, these trials suggest that in the common clinical practice, Everolimus should be administered to ccRCC patients after the failure of immunotherapy and at least one line of TKI.
Everolimus was also tested in other RCC subtypes. The RAPTOR phase II trial evaluates the efficacy of Everolimus as a first-line treatment in a cohort of 92 patients with papillary RCC, reporting a median PFS of 4.1 months, which can be considered a clinical benefit since the treatment options for this variant are scarce.Citation34 Everolimus was also compared with Sunitinib in two major studies enrolling patients with papillary, chromophobe, or unclassified RCC: the ASPEN and the ESPN trials.Citation35,Citation36 In both cases Sunitinib reported a better performance in terms of median PFS.
Temsirolimus
Temsirolimus is the second most widely tested Rapalog. Hudes et alCitation37 tested this drug, within a randomized controlled phase III trial, in treatment-naïve mccRCC patients having a poor prognosis (according to prespecified criteria), who were randomized to receive Temsirolimus, Interferon of a combination of the two agents. The Temsirolimus arm performed better than the others, with a median OS of 10.9 months versus 7.3 months in the Interferon arm and 8.4 months in the combination arm; this indeed was the very first (and for a long time, the only) randomized controlled trial which yielded an OS benefit in mccRCC. On the other hand, the ORR was modest (8.6%).
In a subsequent trial,Citation38 Temsirolimus was compared with Sorafenib, in patients that failed the first-line therapy with Sunitinib. While median PFS did not statistically differ between the two groups (4.3 vs 3.9 months), there was an unexpected significant improvement in median OS in the Sorafenib arm (12.3 vs 16.6 months, HR, 1.31; 95% CI, 1.05 to 1.63; two-sided P = 0.01).
Although Temsirolimus was not as widely tested as Everolimus, available data suggest that it could fit in a similar place in the clinical management of mccRCC.
As for uncommon histologies, the CESAR trial compared Sunitinib and Temsirolimus in patients with papillary, chromophobe, medullary or unclassified RCC. Sunitinib outperformed Temsirolimus in terms of mPFS (13.6 vs 9.3 months, HR 1.75, 95% CI, 0.70–4.46).Citation39
Critical Appraisal of mTOR Inhibitors in mRCC
Upon a cursory glance, one of the weakest aspects of Rapalogs seems to be their relatively low ORR. As a matter of fact, patients that experience shrinkage of lesions are less than 10% in all the aforementioned trials.
Interesting data derive from the analysis of those patients. Indeed, Voss et alCitation40 performed an elegant translational study on the tumors of patients that had long-lasting responses to Rapalogs (median duration: 28 months) and found genomic alterations with activating effect on mTOR signaling in the vast majority of them. Although it can fuel the idea that mTOR mutations could have a predictive role, this study has its limit in the low numerosity of the population and in its retrospective design. More recently, Roldan-Romero et alCitation41 performed genomic and immunohistochemical analysis on a wider cohort of patients that received Rapalogs. Their findings confirmed that mTOR pathway mutations and PTEN loss were significant predictors of partial responses. This data was confirmed also by Kwiatkowski et alCitation42 that studied a population of 79 mRCC patients treated with Rapalogs. Mutations in mTOR and its client network resulted to be more frequent among responders than non-responders. This evidence corroborates the idea that a strict population of Rapalogs responders exists among patients with mRCC, and that it could be selected by finding the correct biomarker. However, it should be noted that all these studies are flawed by their retrospective design.
The only known prospective clinical trial evaluating this aspect, was recently completed by Adib et al.Citation43 In this histology-agnostic trial, a pan-cancer cohort of patients with mTOR mutations was treated with Everolimus, obtaining a disappointing ORR of 7%. Therefore, even if mTOR mutations could have a role in the prediction of response to Everolimus, it is likely that they are not the only factor in the equation.
Nevertheless, even if mTOR mutations were confirmed to be the perfect biomarker, Rapalogs are still flawed by their safety profile. Common adverse events include hematological complications (thrombocytopenia, leukopenia, neutropenia, lymphopenia, and anemia), nausea, mucositis, diarrhea.Citation44 Beyond these adverse events, which are common across different anticancer agents, one of the most feared complications associated with the use of mTOR inhibitors is noninfectious pneumonitis, which can result in acute respiratory distress. In a randomized trialCitation45 comparing Everolimus vs placebo in patients with mccRCC, 16% developed grade 2 and 3.6% grade 3 noninfectious pneumonitis in the Everolimus group.
In conclusion, in mRCC Rapalogs are drugs endowed with a relative and unpredictable efficacy, and an often challenging safety profile. Given the premise of their extraordinary in-vitro results, their clinical performance was disappointing. The reasons behind this debacle have to be researched in the rapid development of resistance, a complex phenomenon, that arises from multiple fronts.
Mechanisms of Resistance to mTOR Inhibitors
The main pathways and mechanisms implied in resistance to mTOR inhibitors are displayed in .
Figure 1 Main mechanisms involved in resistance to Rapalogs.
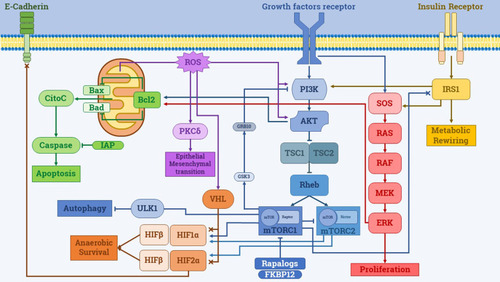
PI3K/AKT
As stated before, Rapalogs bind FKBP12, leading to the inactivation of pivotal proteins of the mTORC1 complex, while mTORC2 remains unaffected.Citation46 It has been demonstrated that mTORC1 inhibition leads to the reduction of Rictor (mTORC2 functional repressor) phosphorylation.Citation47 This will cause disinhibition of mTORC2, with an increase in its activity and a consequential hyperactivation of AKT.Citation48
Moreover, the mTORC1 signal is able to activate, in a GSK3 (glycogen synthase kinase 3)-dependent way, the adapter protein GRB10, which mediates the PI3K-AKT negative feedback.Citation49 It follows that mTOR inhibition would lead to a loss of the PI3K-AKT negative feedback.
In addition, mTORC1 is indirectly responsible for the phosphorylation and subsequent degradation of IRS1 (insulin receptor substrate 1), which has an important role in facilitating insulin signaling,Citation50 which is implied in many anabolic mechanisms. It’s therefore reasonable to hypothesize that the inhibition of mTORC1 caused by Rapalogs will disrupt the PI3K-AKT negative feedback and it will amplify the effects of insulin receptor signaling, thus leading to the activation of the MAP kinases and increasing cell proliferation, angiogenesis, survival, and a metabolic rewiring directed toward anaerobic glycolysis. This could be one of the reasons behind the poor performance of this drug class.
Hypoxia Inducible Factor
HIF is strongly related to RCC tumorigenesis.Citation51 Under normal oxygen concentrations, the subunit HIF-1α and HIF-2α are constantly degraded by a VHL-dependent mechanism involving the proteasome.Citation52 In many cases, however, RCC is characterized by a loss of VHL,Citation53 resulting in persistent activation of HIF. In this scenario, HIF-α subunits will be able to enter the nucleus even if a normal concentration of oxygen exists, and it will bind HIF-β, thus promoting the expression of genes implied in cell survival, apoptosis inhibition, and anaerobic metabolic rewiring.Citation54 However, it has been demonstrated that, while HIF-1α can exert both pro-proliferative and anti-proliferative effects, HIF-2α has only pro-proliferative effects.Citation55 Moreover, HIF-2α is always overexpressed in VHL-defective tumors, while HIF-1α lacks in one-third of them.Citation56
Interestingly, HIF-α subunits translation is stimulated by the PI3K/ATK/mTOR pathway.Citation48 In particular, while HIF-1α expression depends on both mTORC1 and mTORC2, HIF-2α depends only on mTORC2.Citation57 This data suggests that Rapalogs inhibition, which focuses on mTORC1, will not affect HIF-mediated RCC cells’ survival improvement. Furthermore, it has been shown that E-cadherin is a target of the mTORC2/HIF-2α axis. In particular, E-Cadherin is down-regulated by mTORC2/ HIF-2α, leading to the loss of contact inhibition and promoting cell migration and epithelial-to-mesenchymal transition.Citation58
Bcl2/IAP and Apoptosis Evasion
The antiapoptotic protein Bcl2 acts by regulating the permeability of the mitochondrial membrane and the cytoplasmatic release of cytochrome c. The role of Bcl2 in inhibiting apoptosis induced by Rapalogs is well-known and it has been extensively described.Citation59
Another mechanism involves Survivin, a member of the IAP family, that acts by directly inhibiting caspases. There is indirect evidence that Survivin plays an important role in the evasion Rapalogs mediated apoptosis. It has been demonstrated that Rapalogs-mediated apoptosis can be greatly enhanced by reducing Survivin levels.Citation60
Mitogen-Activated Protein Kinases (MAPK) Pathway
MAP kinases are a family of intracellular kinases involved in proliferation, differentiation, mitosis, cell survival and apoptosis. Among MAPK c-Jun N-terminal kinases (JNKs) and p38 MAPKs (p38s) are usually triggered by cellular stresses and inflammatory cytokines signaling, while extracellular signal-regulated kinases (ERKs) are stimulated by growth factors.Citation61
Inhibition of mTOR was demonstrated to induce an activation of the MAP-kinase ERK via a PI3K-dependent feedback loop. In particular, strong activation of the MAPK signaling was observed in vitro, after the administration of Everolimus. In the same study, the inhibition of the MAP-kinase MEK led to the increased anticancer activity of mTOR inhibition both in vitro and in vivo.Citation62
Reactive Oxygen Species (ROS)
ROS plays a major role in several steps of tumorigenesis.Citation63 In VHL-defective RCC, ROS production is enhanced because of the impairment of phosphorylative oxidation.Citation64 It has been demonstrated that PI3K and AKT show increased activity when ROS concentration rises. This response ensures cell survival in stressful conditions.Citation65
Another mechanism involves protein kinase C (PKC) family members, which are mTORC2 downstream effectors.Citation66 The effects of PKC include cell proliferation and epithelial-to-mesenchymal transition by modulation of integrin expression.Citation67 ROS are able to activate different members of the PKC family. In particular, PKCδ is activated by ROS in an Src-dependent mechanism that leads to the activation of Jun MAPK.Citation66
Finally, it was shown that in RCC, a high concentration of ROS can reduce the degradation of HIF-1α,Citation68 thus activating the aforementioned mechanisms that lead to Rapalogs resistance and cell survival. It appears clear that ROS represent an important mechanism to bypass mTOR inhibition since they can activate directly downstream effectors of the mTOR pathway.
AutophagymTOR is considered the principal regulator of the process known as autophagy. Autophagy consists of a mechanism that cells use to recycle damaged organelles in order to recover nutrients and sustain energy production in stressful conditions. It has been demonstrated that mTOR acts as a nutrient sensor, that stimulates cell proliferation, and shuts down autophagy in a ULK1-dependent fashion.Citation69 Therefore, mTOR blockade acts as a stimulus for autophagy, that causes an increase in nutrients availability to sustain energy production, thus increasing the survival capability of cancer cells.Citation70 As a counter-proof, available data suggested that the inhibition of the autophagic machinery key component Atg7 can enhance the anti-cancer activity of Rapalogs.Citation71 From a mechanistic point of view, this process is explained by the impairment in phosphorylative oxidation caused by mTOR inhibition, which leads to an increase in ROS concentration and mitochondrial damage. Damaged mitochondria are usually destroyed via autophagy, but if Atg7 is inhibited, they continue to produce ROS up to the point at which oxidative stress causes cell death. Interestingly, this form of death is not caspase-dependent, being not apoptosis, but rather a form of necroptosis.Citation71
Immune Response Impairment
Rapalogs are primarily considered to be immunosuppressive drugs. Treatment with mTOR inhibitors induces the development of T regulatory cells and makes them more resistant to apoptosis.Citation72 RCC is an immunologically “hot” tumor and the growing role of immunotherapy in its treatment can be considered proof of the importance of an active immune system in the management of this disease.Citation73 Data show that in patients treated with Everolimus, the worst prognosis has been demonstrated when the neutrophil to lymphocyte ratio is higher than 3.Citation74 This is an intrinsic flaw of Rapalogs since their immunosuppressive effect cannot be dissociated from their anticancer effect. However, the relative strength of immune suppression could theoretically be related to the dose used; indeed, the dose commonly administered to transplanted patients for immunosuppressive purposes is completely different from that used in the oncological setting.
On the other hand, mTOR inhibitors could be a suitable option for mRCC in kidney transplant patients, since they could exert both the immunosuppressive and the anti-cancer effect.Citation75
Overcoming the Resistance to mTOR Inhibitors
The principal clinical trials exploiting strategies to overcome resistance to mTOR inhibitors are summarized in .
Table 1 Main Clinical Trials Featuring Strategies to Overcome mTOR Inhibition
Second and Third-Generation mTOR Inhibitors
The most immediate strategy to overcome the aforementioned resistance mechanism is the development of more potent mTOR inhibitors. Second-generation mTOR inhibitors, TOR Kinase Inhibitors (TOR-KI), are able to block mTOR in its catalytic domain, by competing with ATP. TOR-KIs do not need FKBP to exert their action since they are developed to bypass this step. The key feature of TOR-KIs, is their ability to block both mTORC1 and mTORC2, thus overcoming some of the aforementioned described mechanisms of resistance. In fact, TOR-KIs were proven to be effective in Rapamycin-insensitive cell lines.Citation76 However, resistance remains a major concern, since they are not so effective at preventing the feedback activation of PI3K that implies GSK3 and IRS-1.Citation77
Vistusertib (AZD2014) was the first molecule of this class to reach clinical experimentation. A phase II clinical trial compared Vistusertib with Everolimus in patients with mccRCC beyond the first line of treatment.Citation78 The trial was stopped before the completion of the enrollment because of the lack of efficacy observed in Vistusertib’s arm. PFS greatly favored Everolimus (1.6 vs 4.8 months, p<0.01). The only true advantage offered by Vistusertib was a slightly more manageable safety profile.
Another TOR-KIs that reached human testing is Sapanisertib (TAK-228).Citation79 A Phase I dose-finding study demonstrated a good safety profile for this drug and an ORR of 22% was observed in one of the cohorts. More clinical trials are needed to point out a possible clinical role for this class of molecules.
Third-generation mTOR inhibitors are currently in their preclinical phase. They were obtained by connecting a first-generation mTOR inhibitor and a second-generation TOR Kinase Inhibitor through a linker.Citation80 RapaLink-1 is the first compound of this new class, and it is composed of Rapamycin linked to the TOR-KI MLN0128. It was recently compared with Temsirolimus in RCC cell lines, showing a greater anti-tumor efficacy both in Sunitinib-resistant and Sunitinib-sensitive cell lines.Citation81
The development of these novel inhibitors could improve the performances observed from the experience with Rapalogs. However, many of the resistance mechanisms described do not depend on the potency of inhibitors per se, but rather on a complex interplay between mTOR and other pathways. Hence the consideration that drugging more than just one molecular target could offer sensible improvements.
Combination of VEGFR Inhibitors and mTOR Inhibitors
The idea of combining mTOR inhibitors and VEGFR-inhibitors arose soon after the first results of trials with antiangiogenics. Blocking both the angiogenesis and mTOR pathway could deliver enough metabolic damage to RCC cells to improve clinical outcomes and delay the onset of resistance. The first phase I study exploiting this combination was a dose-finding study evaluating the safety of Sunitinib and Everolimus in pretreated mccRCC patients.Citation82 Acute and chronic grade 3 and 4 toxicities were reported among all the cohorts, and the combination resulted to be tolerated only at attenuated doses. The unmanageable safety profile of this combination was confirmed also by a subsequent phase Ib trial that reported similar results.Citation83
Another combination explored was Lenvatinib and Everolimus. In a phase II trial,Citation84 153 mccRCC patients that failed previous antiangiogenic therapy, were randomized to receive either Lenvatinib, Everolimus, or both in combination. The combination arm reported a longer mPFS than both the monotherapy arms (14.6 months vs 5.5 months for Everolimus and 7.4 months of Lenvatinib alone), although the data were statistically significant only in the comparison with the Everolimus arm (HR 0.40, 95% CI 0.24–0.68; p=0.0005). Moreover, the combination arm reported more grade 3 and 4 adverse events, but in the end, the combination therapy showed an acceptable safety profile.
This combination was also evaluated in a phase III trial involving naïve mccRCC patients that were randomized to receive Pembrolizumab plus Lenvatinib, Everolimus plus Lenvatinib or Sunitinib.Citation85 The Pembrolizumab-Lenvatinib arm performed much better than Sunitinib in terms of mPFS (23.9 months vs 9.2 months; p<0.001), but also the Everolimus-Lenvatinib arm reported a good outcome (14.7 months of mPFS). Both the combination arms showed a slightly higher frequency of adverse events, but they had an overall manageable safety profile. The results reported in these studies led to the design of a clinical trial exploiting Lenvatinib plus Everolimus as a neoadjuvant treatment aimed at making unresectable RCC resectable This trial is currently ongoing (NCT03324373).
The combination of Lenvatinib and Everolimus was tested also in a phase II trial enrolling a cohort of 31 patients with uncommon RCC subtypes reporting a ORR of 25.8% and a DCR of 61.3%, with a mPFS of 9.2 months.Citation86 This is an important milestone for the treatment of RCC variants that often lack of valuable therapeutic options.
Some better results come from a phase Ib trial of the VEGFR-inhibitor Vatalanib in combination with Everolimus.Citation87 Even though adverse events were observed, long-term tolerability was demonstrated in a significant portion of patients. Moreover, an mOS of 16.3 months was reported with an ORR of 29.2%. Interestingly, the mOS was much longer for treatment-naïve patients (25.1 months). These data suggest that the combination of Valatanib and Everolimus could be more effective than the sequential schedule at obtaining long-lasting responses. More recently, another phase I trial analyzed this combination in a cohort of metastatic solid tumors, and the data were confirmed in the mccRCC subgroup.Citation88
Similar results were reported by a phase I trial investigating the combination of the VEGFR-inhibitor Vorolanib (X-82, CM082) and Everolimus in mccRCC pretreated patients.Citation89 Along with the good tolerability of the treatment, a good efficacy emerged from this study, with an ORR of 32% and a DCR of 100%. These data are confirmed also by another phase I trial exploiting this combination in solid tumors: the mccRCC cohort was the one with the best ORR.Citation90
In the end, the combination of VEGFR inhibitors and mTOR inhibitors resulted to be difficult to manage in some cases but reported an efficacy profile good enough to prompt further investigation.
Alternating VEGFR Inhibitors and mTOR Inhibitors
A theoretically simple strategy to avoid the toxicities of the combination, while reducing the resistance phenomena, could be the sequential alternation of VEGFR inhibition and mTOR inhibition. The rationale of this therapeutic schedule resides in the hypothesis that VEGFR-inhibitors would select a clone of RCC cells that are sensitive to mTOR inhibition, and vice-versa. The EVERSUN phase II trial tested an alternating schedule of Sunitinib and Everolimus in 55 naïve mccRCC patients, obtaining a median PFS of 8 months and a median OS of 17 months.Citation91 These results did not meet the pre-specified values to warrant further research.
In the ROPETAR trial, 101 naïve mccRCC patients were randomized to receive either an alternating schedule of Pazopanib and Everolimus or continuous Pazopanib.Citation92 The Pazopanib arm performed better in terms of median PFS (9.4 vs 7.4 months) and no significant differences were observed in the safety profile, thus suggesting that the rotating schedule gives no advantages at all.
Similar findings were reported by the SUNRISE study, a phase II trial comparing alternating cycles of Sunitinib and Everolimus versus standard sequential treatment of Sunitinib followed by Everolimus upon progression, in 42 naïve mccRCC patients.Citation93 The 1-year PFS rate was 49.7% in the alternating arm and 84.62% in the sequential arm, with no major differences in mOS. From this data, it can be inferred that the alternating strategy fails to improve the clinical outcomes in mccRCC. The almost contemporary exposure to both drugs likely favors the development of resistance mechanisms allowing mccRCC cells to acquire resistance to both VEGFR-inhibition and mTOR-inhibition.
PI3K/mTOR Dual Inhibitors
Among all the resistance mechanisms analyzed, the PI3K feedback is surely one of the most common, and its onset cannot be delayed by simply increasing the potency of mTOR blockade. Hence the idea of developing molecules that can block both mTOR and PI3K.
One of the first clinical studies involving this new class of drugs was a phase Ib trial of BEZ235 previously treated patients with mccRCC.Citation94 Even though BEZ235 was extensively tested in the preclinical setting, the trial was interrupted due to the high incidence of dose-limiting toxicities including fatigue, rash, nausea and vomiting, diarrhea, mucositis, anorexia, and dysgeusia. Therefore, the development of the drug was stopped.
Another drug of this class, Apitolisib (GDC-0980) was compared with Everolimus in a phase II clinical trial enrolling mccRCC patients that progressed on after the first-line treatment with VEGFR-inhibitors.Citation95 Surprisingly, the Everolimus arm had a longer median PFS (6.1 vs 3.7 months) and median OS (22.8 vs 16.5 months), and a higher ORR (11.6% vs 7.1%). The authors explained these results with the higher frequency of adverse events observed in the Apitolisib arm. A possible explication for the unexpected bad performance of PI3K/mTOR dual inhibitors could be that the resistance mechanisms implying PI3K, although frequent, are not pivotal for cell survival. Moreover, the unfavorable safety profile of these drugs makes it difficult to evaluate objectively their results. Finally, it should be pointed out that the design of this trial could have been improved. In fact, Apitolisib could have been administered to patients progressed beyond a first line therapy with anti-angiogenics and a second line therapy with Everolimus. In this setting, Apitolisib should have been compared with best supportive care or clinician choice, thus demonstrating if it is effectively capable of overcoming the resistance built by Everolimus through its innovative mechanism of action.
Combination of mTOR Inhibitors and Histone Deacetylase (HDAC) Inhibitors
The inhibition of HDAC exerts its anticancer effect by modulating DNA coiling and thus, genes expression. Preclinical data showed that AKT phosphorylation is negatively affected by HDAC inhibition, although the precise mechanism of the phenomenon remains unclear.Citation96 Hence the rationale for combination therapy.
A recent phase I clinical trial tested the rapalog Ridaforolimus with the HDAC inhibitor Vorinostat in mccRCC pretreated patients.Citation97 Although it was only a dose-finding study, some of the patients experimented a long-lasting disease stabilization, and no safety concerns were observed. Similar results were observed in another phase I trial with the combination of Everolimus and the HDAC inhibitor Panobinostat.Citation98 Moreover, this combination reported a 4.1 months median PFS, although more standardized studies involving larger populations are needed to clarify the possible role of this combination in mRCC therapy.
Combination of Autophagy Inhibitors and mTOR Inhibitors
As discussed before, mTOR blockade promotes autophagy processes that cancer cells use to recycle damaged organelles and produce energy. The drug Hydroxychloroquine is known for its ability to inhibit autophagy, by limiting the fusion of lysosomes and autophagosomes.Citation99 Haas et al studied the efficacy and safety of combining Hydroxychloroquine and Everolimus in a cohort of 33 patients that had already failed previous therapy lines. Disease control was achieved in 66% of patients, with an ORR of 6%. No dose-limiting toxicity was reported. Interestingly, this phase I/II trial met its primary endpoint, with a 6-months-PFS higher than 40%.Citation100 The data shown here confirm the importance of autophagy in the onset of resistance to rapalogs, but further investigation is necessary to understand exactly the extent of the improvement offered by autophagy inhibitors.
Conclusion
Despite the rational of targeting mTOR in mRCC, and its pivotal role in RCC tumorigenesis, the inhibition of mTOR through Rapalogs did not meet the expectations. Objective responses were scarce and, more importantly, disease control was brief.
These results are principally attributable to the complex interplay between the mTOR pathway and many other cell growth and proliferation pathways, that allows the onset of resistance phenomena. At the state of the art, some solutions to overcome the resistance are under development. On the one hand, more potent and innovative mTOR inhibitors are being created, to obtain an improved blockade of both mTORC1 and mTORC2. On the other hand, combinatorial strategies aim at countering some of the known resistance mechanisms. In the near future, more combinations of mTOR inhibitors and VEGFR inhibitors could reach clinical practice. More studies are needed for combinations including autophagy blockers and HDAC inhibitors, both of which showed promising preliminary results.Citation97,Citation98,Citation100 Finally, completely unexplored remains the combination of mTOR inhibitors and immune checkpoint inhibitors.
Even if it may appear counterintuitive (potentially immune suppressive agents combined with an immune stimulating one?), this synergy could improve the effectiveness of immune therapy, which has already shown excellent results in mRCC.
Disclosure
Dr Mimma Rizzo reports personal fees from MSD, personal fees from Pfizer, personal fees from Novartis, personal fees from AstraZeneca, during the conduct of the study.
Prof. Dr. Camillo Porta reports personal fees from Angelini Pharma, personal fees from AstraZeneca, personal fees from BMS, personal fees from Eisai, personal fees from EUSA Pharma, personal fees from General Electric, personal fees from Ipsen, personal fees from Janssen, personal fees from Merck Serono, personal fees from MSD, personal fees from Novartis, personal fees from Pfizer, and Roche, outside the submitted work. The authors report no conflicts of interest relative to the present manuscript.
References
- Tsang CK, Qi H, Liu LF, Zheng XF. Targeting mammalian target of rapamycin (mTOR) for health and diseases. Drug Discov Today. 2007;12(3–4):112–124. doi:10.1016/j.drudis.2006.12.00817275731
- Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124(3):471–484. doi:10.1016/j.cell.2006.01.01616469695
- Hudes GR. Targeting mTOR in renal cell carcinoma. Cancer. 2009;115(10):2313–2320. doi:10.1002/cncr.2423919402072
- Watanabe R, Wei L, Huang J. mTOR signaling, function, novel inhibitors, and therapeutic targets. J Nucl Med. 2011;52(4):497–500. doi:10.2967/jnumed.111.08962321421716
- Neshat MS, Mellinghoff IK, Tran C, et al. Enhanced sensitivity of PTEN-deficient tumors to inhibition of FRAP/mTOR. Proc Natl Acad Sci USA. 2001;98(18):10314–10319. doi:10.1073/pnas.17107679811504908
- Podsypanina K, Lee RT, Politis C, et al. An inhibitor of mTOR reduces neoplasia and normalizes p70/S6 kinase activity in Pten ± mice. Proc Natl Acad Sci USA. 2001;98(18):10320–10325. doi:10.1073/pnas.17106009811504907
- Sarbassov DD, Ali SM, Sabatini DM. Growing roles for the mTOR pathway. Curr Opin Cell Biol. 2005;17(6):596–603. doi:10.1016/j.ceb.2005.09.00916226444
- Vivanco I, Sawyers CL. The phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2(7):489–501. doi:10.1038/nrc83912094235
- Sato T, Nakashima A, Guo L, Coffman K, Tamanoi F. Single amino-acid changes that confer constitutive activation of mTOR are discovered in human cancer. Oncogene. 2010;29(18):2746–2752. doi:10.1038/onc.2010.2820190810
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307(5712):1098–1101. doi:10.1126/science.110614815718470
- Ma L, Teruya-Feldstein J, Behrendt N, et al. Genetic analysis of Pten and Tsc2 functional interactions in the mouse reveals asymmetrical haploinsufficiency in tumor suppression. Genes Dev. 2005;19(15):1779–1786. doi:10.1101/gad.131440516027168
- Manning BD, Logsdon MN, Lipovsky AI, Abbott D, Kwiatkowski DJ, Cantley LC. Feedback inhibition of Akt signaling limits the growth of tumors lacking Tsc2. Genes Dev. 2005;19(15):1773–1778. doi:10.1101/gad.131460516027169
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499(7456):43–49. doi:10.1038/nature1222223792563
- Hager M, Haufe H, Lusuardi L, Schmeller N, Kolbitsch C. PTEN, pAKT, and pmTOR expression and subcellular distribution in primary renal cell carcinomas and their metastases. Cancer Invest. 2011;29(7):427–438. doi:10.3109/07357907.2011.58478221696297
- Cargnello M, Tcherkezian J, Roux PP. The expanding role of mTOR in cancer cell growth and proliferation. Mutagenesis. 2015;30(2):169–176. doi:10.1093/mutage/geu04525688110
- Guo H, German P, Bai S, et al. The PI3K/AKT pathway and renal cell carcinoma. J Genet Genomics. 2015;42(7):343–353. doi:10.1016/j.jgg.2015.03.00326233890
- Han G, Zhao W, Song X, et al. Unique protein expression signatures of survival time in kidney renal clear cell carcinoma through a pan-cancer screening. BMC Genomics. 2017;18(6):678. doi:10.1186/s12864-017-4026-628984208
- Zoumpourlis P, Genovese G, Tannir NM, Msaouel P. Systemic therapies for the management of non-clear cell renal cell carcinoma: what works, what doesn’t, and what the future holds. Clin Genitourin Cancer. 2021;19(2):103–116. doi:10.1016/j.clgc.2020.11.00533358151
- Trpkov K, Williamson SR, Gill AJ, et al. Novel, emerging and provisional renal entities: the Genitourinary Pathology Society (GUPS) update on renal neoplasia. Mod Pathol. 2021;34(6):1167–1184. doi:10.1038/s41379-021-00737-633526874
- Fan D, Liu Q, Wu F, et al. Prognostic significance of PI3K/AKT/ mTOR signaling pathway members in clear cell renal cell carcinoma. PeerJ. 2020;8:e9261. doi:10.7717/peerj.926132547875
- Ocana A, Vera-Badillo F, Al-Mubarak M, et al. Activation of the PI3K/mTOR/AKT pathway and survival in solid tumors: systematic review and meta-analysis. PLoS One. 2014;9(4):e95219. doi:10.1371/journal.pone.009521924777052
- Jonasch E, Gao J, Rathmell WK. Renal cell carcinoma. BMJ. 2014;349:g4797. doi:10.1136/bmj.g479725385470
- Linehan WM, Rathmell WK. Kidney cancer. Urol Oncol. 2012;30(6):948–951. doi:10.1016/j.urolonc.2012.08.02123218074
- Dumont FJ, Su Q. Mechanism of action of the immunosuppressant rapamycin. Life Sci. 1996;58(5):373–395. doi:10.1016/0024-3205(95)02233-38594303
- Escudier B, Porta C, Bono P, et al. Randomized, controlled, double-blind, cross-over trial assessing treatment preference for pazopanib versus sunitinib in patients with metastatic renal cell carcinoma: PISCES Study. J Clin Oncol. 2014;32(14):1412–1418. doi:10.1200/JCO.2013.50.826724687826
- Ghidini M, Petrelli F, Ghidini A, et al. Clinical development of mTor inhibitors for renal cancer. Expert Opin Investig Drugs. 2017;26(11):1229–1237. doi:10.1080/13543784.2017.1384813
- Luan FL, Ding R, Sharma VK, Chon WJ, Lagman M, Suthanthiran M. Rapamycin is an effective inhibitor of human renal cancer metastasis. Kidney Int. 2003;63(3):917–926. doi:10.1046/j.1523-1755.2003.00805.x12631072
- Amato RJ, Jac J, Giessinger S, Saxena S, Willis JP. A Phase 2 study with a daily regimen of the oral mTOR inhibitor RAD001 (everolimus) in patients with metastatic clear cell renal cell cancer. Cancer. 2009;115(11):2438–2446. doi:10.1002/cncr.2428019306412
- Motzer RJ, Escudier B, Oudard S, et al. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet. 2008;372(9637):449–456. doi:10.1016/S0140-6736(08)61039-918653228
- Ravaud A, Barrios CH, Alekseev B, et al. RECORD-2: phase II randomized study of everolimus and bevacizumab versus interferon α-2a and bevacizumab as first-line therapy in patients with metastatic renal cell carcinoma. Ann Oncol. 2015;26(7):1378–1384. doi:10.1093/annonc/mdv17025851632
- Motzer RJ, Barrios CH, Kim TM, et al. Phase II randomized trial comparing sequential first-line everolimus and second-line sunitinib versus first-line sunitinib and second-line everolimus in patients with metastatic renal cell carcinoma. J Clin Oncol. 2014;32(25):2765–2772. doi:10.1200/JCO.2013.54.691125049330
- Choueiri TK, Escudier B, Powles T, et al. Cabozantinib versus everolimus in advanced renal cell carcinoma (METEOR): final results from a randomised, open-label, Phase 3 trial. Lancet Oncol. 2016;17(7):917–927. doi:10.1016/S1470-2045(16)30107-327279544
- Motzer RJ, Escudier B, George S, et al. Nivolumab versus everolimus in patients with advanced renal cell carcinoma: updated results with long-term follow-up of the randomized, open-label, phase 3 CheckMate 025 trial. Cancer. 2020;126(18):4156–4167. doi:10.1002/cncr.3303332673417
- Escudier B, Molinie V, Bracarda S, et al. Open-label phase 2 trial of first-line everolimus monotherapy in patients with papillary metastatic renal cell carcinoma: RAPTOR final analysis. Eur J Cancer. 2016;69:226–235. doi:10.1016/j.ejca.2016.08.00427680407
- Armstrong AJ, Halabi S, Eisen T, et al. Everolimus versus sunitinib for patients with metastatic non-clear cell renal cell carcinoma (ASPEN): a multicentre, open-label, randomised phase 2 trial. Lancet Oncol. 2016;17(3):378–388. doi:10.1016/S1470-2045(15)00515-X26794930
- Tannir NM, Jonasch E, Albiges L, et al. Everolimus versus sunitinib prospective evaluation in metastatic non-clear cell renal cell carcinoma (ESPN): a randomized multicenter phase 2 trial. Eur Urol. 2016;69(5):866–874. doi:10.1016/j.eururo.2015.10.04926626617
- Hudes G, Carducci M, Tomczak P, et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med. 2007;356(22):2271–2281. doi:10.1056/NEJMoa06683817538086
- Hutson TE, Escudier B, Esteban E, et al. Randomized phase III trial of temsirolimus versus sorafenib as second-line therapy after sunitinib in patients with metastatic renal cell carcinoma. J Clin Oncol. 2014;32(8):760–767. doi:10.1200/JCO.2013.50.396124297950
- Bergmann L, Grünwald V, Maute L, et al. A randomized phase iia trial with temsirolimus versus sunitinib in advanced non-clear cell renal cell carcinoma: an intergroup study of the CESAR Central European Society for Anticancer Drug Research-EWIV and the Interdisciplinary Working Group on renal cell cancer (IAGN) of the German cancer society. Oncol Res Treat. 2020;43(7–8):333–339.32541143
- Voss MH, Hakimi AA, Pham CG, et al. Tumor genetic analyses of patients with metastatic renal cell carcinoma and extended benefit from mTOR inhibitor therapy. Clin Cancer Res. 2014;20(7):1955–1964. doi:10.1158/1078-0432.CCR-13-234524622468
- Roldan-Romero JM, Beuselinck B, Santos M, et al. PTEN expression and mutations in TSC1, TSC2 and MTOR are associated with response to rapalogs in patients with renal cell carcinoma. Int J Cancer. 2020;146(5):1435–1444. doi:10.1002/ijc.3257931335987
- Kwiatkowski DJ, Choueiri TK, Fay AP, et al. Mutations in TSC1, TSC2, and MTOR are associated with response to rapalogs in patients with metastatic renal cell carcinoma. Clin Cancer Res. 2016;22(10):2445–2452. doi:10.1158/1078-0432.CCR-15-263126831717
- Adib E, Klonowska K, Giannikou K, et al. Phase II clinical trial of everolimus in a pan-cancer cohort of patients with mTOR pathway alterations. Clin Cancer Res. 2021;27(14):3845–3853. doi:10.1158/1078-0432.CCR-20-454833727259
- Basu B, Dean E, Puglisi M, et al. First-in-human pharmacokinetic and pharmacodynamic study of the dual m-TORC 1/2 inhibitor AZD2014. Clin Cancer Res. 2015;21(15):3412–3419. doi:10.1158/1078-0432.CCR-14-242225805799
- White DA, Camus P, Endo M, et al. Noninfectious pneumonitis after everolimus therapy for advanced renal cell carcinoma. Am J Respir Crit Care Med. 2010;182(3):396–403. doi:10.1164/rccm.200911-1720OC20194812
- Sarbassov DD, Ali SM, Sengupta S, et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22(2):159–168. doi:10.1016/j.molcel.2006.03.02916603397
- Dibble CC, Asara JM, Manning BD. Characterization of Rictor phosphorylation sites reveals direct regulation of mTOR complex 2 by S6K1. Mol Cell Biol. 2009;29(21):5657–5670. doi:10.1128/MCB.00735-0919720745
- O’Reilly KE, Rojo F, She QB, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66(3):1500–1508. doi:10.1158/0008-5472.CAN-05-292516452206
- Yu Y, Yoon SO, Poulogiannis G, et al. Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science. 2011;332(6035):1322–1326. doi:10.1126/science.119948421659605
- Harrington LS, Findlay GM, Gray A, et al. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J Cell Biol. 2004;166(2):213–223. doi:10.1083/jcb.20040306915249583
- Martínez-Sáez O, Gajate Borau P, Alonso-Gordoa T, Molina-Cerrillo J, Grande E. Targeting HIF-2 α in clear cell renal cell carcinoma: a promising therapeutic strategy. Crit Rev Oncol Hematol. 2017;111:117–123. doi:10.1016/j.critrevonc.2017.01.01328259286
- Ohh M, Park CW, Ivan M, et al. Ubiquitination of hypoxia-inducible factor requires direct binding to the β-domain of the von Hippel–Lindau protein. Nat Cell Biol. 2000;2(7):423–427. doi:10.1038/3501705410878807
- Linehan WM, Rubin JS, Bottaro DP. VHL loss of function and its impact on oncogenic signaling networks in clear cell renal cell carcinoma. Int J Biochem Cell Biol. 2009;41(4):753–756. doi:10.1016/j.biocel.2008.09.02418950731
- Toschi A, Lee E, Gadir N, Ohh M, Foster DA. Differential dependence of hypoxia-inducible factors 1α and 2α on mTORC1 and mTORC2. J Biol Chem. 2008;283(50):34495–34499. doi:10.1074/jbc.C80017020018945681
- Kondo K, Kim WY, Lechpammer M, Kaelin WG Jr. Inhibition of HIF2α is sufficient to suppress pVHL-defective tumor growth. PLoS Biol. 2003;1(3):E83. doi:10.1371/journal.pbio.000008314691554
- Maranchie JK, Vasselli JR, Riss J, Bonifacino JS, Linehan WM, Klausner RD. The contribution of VHL substrate binding and HIF1-α to the phenotype of VHL loss in renal cell carcinoma. Cancer Cell. 2002;1(3):247–255. doi:10.1016/S1535-6108(02)00044-212086861
- Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12(1):9–22. doi:10.1016/j.ccr.2007.05.00817613433
- Evans AJ, Russell RC, Roche O, et al. VHL promotes E2 box-dependent E-cadherin transcription by HIF-mediated regulation of SIP1 and snail. Mol Cell Biol. 2007;27(1):157–169. doi:10.1128/MCB.00892-0617060462
- Hosoi H, Dilling MB, Shikata T, et al. Rapamycin causes poorly reversible inhibition of mTOR and induces p53-independent apoptosis in human rhabdomyosarcoma cells. Cancer Res. 1999;59(4):886–894.10029080
- Mahalingam D, Medina EC, Esquivel JA, et al. Vorinostat enhances the activity of temsirolimus in renal cell carcinoma through suppression of survivin levels. Clin Cancer Res. 2010;16(1):141–153. doi:10.1158/1078-0432.CCR-09-138520028765
- Pearson G, Robinson F, Beers Gibson T, et al. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev. 2001;22(2):153–183.11294822
- Carracedo A, Ma L, Teruya-Feldstein J, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest. 2008;118(9):3065–3074.18725988
- Kirtonia A, Sethi G, Garg M. The multifaceted role of reactive oxygen species in tumorigenesis. Cell Mol Life Sci. 2020;77(22):4459–4483.32358622
- Hervouet E, Simonnet H, Godinot C. Mitochondria and reactive oxygen species in renal cancer. Biochimie. 2007;89(9):1080–1088. doi:10.1016/j.biochi.2007.03.01017466430
- Okoh VO, Felty Q, Parkash J, Poppiti R, Roy D. Reactive oxygen species via redox signaling to PI3K/AKT pathway contribute to the malignant growth of 4-hydroxy estradiol-transformed mammary epithelial cells. PLoS One. 2013;8(2):e54206. doi:10.1371/journal.pone.005420623437041
- Basu A, Sridharan S, Persaud S. Regulation of protein kinase C delta downregulation by protein kinase C epsilon and mammalian target of rapamycin complex 2. Cell Signal. 2009;21(11):1680–1685. doi:10.1016/j.cellsig.2009.07.00619632318
- Wu WS, Tsai RK, Chang CH, Wang S, Wu JR, Chang YX. Reactive oxygen species mediated sustained activation of protein kinase C alpha and extracellular signal-regulated kinase for migration of human hepatoma cell Hepg2. Mol Cancer Res. 2006;4(10):747–758. doi:10.1158/1541-7786.MCR-06-009617050668
- Hervouet E, Cízková A, Demont J, et al. HIF and reactive oxygen species regulate oxidative phosphorylation in cancer. Carcinogenesis. 2008;29(8):1528–1537. doi:10.1093/carcin/bgn12518515279
- Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol. 2010;22(2):132–139. doi:10.1016/j.ceb.2009.12.00420056399
- Jung CH, Jun CB, Ro SH, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell. 2009;20(7):1992–2003. doi:10.1091/mbc.e08-12-124919225151
- Bray K, Mathew R, Lau A, et al. Autophagy suppresses RIP kinase-dependent necrosis enabling survival to mTOR inhibition. PLoS One. 2012;7(7):e41831. doi:10.1371/journal.pone.004183122848625
- Strauss L, Czystowska M, Szajnik M, Mandapathil M, Whiteside TL. Differential responses of human regulatory T cells (Treg) and effector T cells to rapamycin. PLoS One. 2009;4(6):e5994. doi:10.1371/journal.pone.000599419543393
- Deleuze A, Saout J, Dugay F, et al. Immunotherapy in renal cell carcinoma: the future is now. Int J Mol Sci. 2020;21(7):2532. doi:10.3390/ijms21072532
- Santoni M, De Giorgi U, Iacovelli R, et al. Pre-treatment neutrophil-to-lymphocyte ratio may be associated with the outcome in patients treated with everolimus for metastatic renal cell carcinoma. Br J Cancer. 2013;109(7):1755–1759. doi:10.1038/bjc.2013.52224008663
- Holdaas H, De Simone P, Zuckermann A. Everolimus and malignancy after solid organ transplantation: a clinical update. J Transplant. 2016;2016:4369574. doi:10.1155/2016/436957427807479
- Feldman ME, Shokat KM. New inhibitors of the PI3K-Akt-mTOR pathway: insights into mTOR signaling from a new generation of Tor Kinase Domain Inhibitors (TORKinibs). Curr Top Microbiol Immunol. 2010;347:241–262.20549474
- Feldman ME, Apsel B, Uotila A, et al. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 2009;7(2):e38. doi:10.1371/journal.pbio.100003819209957
- Powles T, Wheater M, Din O, et al. A randomised phase 2 study of AZD2014 versus everolimus in patients with VEGF-refractory metastatic clear cell renal cancer. Eur Urol. 2016;69(3):450–456. doi:10.1016/j.eururo.2015.08.03526364551
- Voss MH, Gordon MS, Mita M, et al. Phase 1 study of mTORC1/2 inhibitor sapanisertib (TAK-228) in advanced solid tumours, with an expansion phase in renal, endometrial or bladder cancer. Br J Cancer. 2020;123(11):1590–1598. doi:10.1038/s41416-020-01041-x32913286
- Rodrik-Outmezguine VS, Okaniwa M, Yao Z, et al. Overcoming mTOR resistance mutations with a new-generation mTOR inhibitor. Nature. 2016;534(7606):272–276. doi:10.1038/nature1796327279227
- Kuroshima K, Yoshino H, Okamura S, et al. Potential new therapy of Rapalink-1, a new generation mammalian target of rapamycin inhibitor, against sunitinib-resistant renal cell carcinoma. Cancer Sci. 2020;111(5):1607–1618. doi:10.1111/cas.1439532232883
- Molina AM, Feldman DR, Voss MH, et al. Phase 1 trial of everolimus plus sunitinib in patients with metastatic renal cell carcinoma. Cancer. 2012;118(7):1868–1876. doi:10.1002/cncr.2642921898375
- Kanesvaran R, Watt K, Turnbull JD, et al. A single-arm phase 1b study of everolimus and sunitinib in patients with advanced renal cell carcinoma. Clin Genitourin Cancer. 2015;13(4):319–327. doi:10.1016/j.clgc.2014.12.01126174223
- Motzer RJ, Hutson TE, Glen H, et al. Lenvatinib, everolimus, and the combination in patients with metastatic renal cell carcinoma: a randomised, phase 2, open-label, multicentre trial. Lancet Oncol. 2015;16(15):1473–1482. doi:10.1016/S1470-2045(15)00290-926482279
- Motzer R, Alekseev B, Rha SY, et al. Lenvatinib plus pembrolizumab or everolimus for advanced renal cell carcinoma. N Engl J Med. 2021;384(14):1289–1300. doi:10.1056/NEJMoa203571633616314
- Hutson TE, Michaelson MD, Kuzel TM, et al. A single-arm, multicenter, phase 2 study of lenvatinib plus everolimus in patients with advanced non-clear cell renal cell carcinoma. Eur Urol. 2021;80(2):162–170. doi:10.1016/j.eururo.2021.03.01533867192
- Bitting RL, Healy P, Creel PA, et al. A phase Ib study of combined VEGFR and mTOR inhibition with vatalanib and everolimus in patients with advanced renal cell carcinoma. Clin Genitourin Cancer. 2014;12(4):241–250. doi:10.1016/j.clgc.2013.11.02024685058
- Zhu M, Molina JR, Dy GK, et al. A phase I study of the VEGFR kinase inhibitor vatalanib in combination with the mTOR inhibitor, everolimus, in patients with advanced solid tumors. Invest New Drugs. 2020;38(6):1755–1762. doi:10.1007/s10637-020-00936-z32328844
- Sheng X, Yan X, Chi Z, et al. Phase 1 trial of vorolanib (CM082) in combination with everolimus in patients with advanced clear-cell renal cell carcinoma. EBioMedicine. 2020;55:102755. doi:10.1016/j.ebiom.2020.10275532335374
- Pedersen KS, Grierson PM, Picus J, et al. Vorolanib (X-82), an oral anti-VEGFR/PDGFR/CSF1R tyrosine kinase inhibitor, with everolimus in solid tumors: results of a phase I study. Invest New Drugs. 2021;39(5):1298–1305. PMID: 33738668. doi:10.1007/s10637-021-01093-733738668
- Davis ID, Long A, Yip S, et al. EVERSUN: a phase 2 trial of alternating sunitinib and everolimus as first-line therapy for advanced renal cell carcinoma. Ann Oncol. 2015;26(6):1118–1123. doi:10.1093/annonc/mdv07825701452
- Cirkel GA, Hamberg P, Sleijfer S, et al. Alternating treatment with pazopanib and everolimus vs continuous pazopanib to delay disease progression in patients with metastatic clear cell renal cell cancer: the ROPETAR randomized clinical trial. JAMA Oncol. 2017;3(4):501–508. doi:10.1001/jamaoncol.2016.520227918762
- Rodriguez-Vida A, Bamias A, Esteban E, et al. Randomised Phase II study comparing alternating cycles of sunitinib and everolimus vs standard sequential administration in first-line metastatic renal carcinoma (SUNRISES study). BJU Int. 2020;126(5):559–567. doi:10.1111/bju.1516532654362
- Carlo MI, Molina AM, Lakhman Y, et al. A phase Ib study of BEZ235, a dual inhibitor of phosphatidylinositol 3-kinase (PI3K) and mammalian target of rapamycin (mTOR), in patients with advanced renal cell carcinoma. Oncologist. 2016;21(7):787–788. doi:10.1634/theoncologist.2016-014527286790
- Powles T, Lackner MR, Oudard S, et al. Randomized open-label phase II trial of apitolisib (GDC-0980), a novel inhibitor of the PI3K/mammalian target of rapamycin pathway, versus everolimus in patients with metastatic renal cell carcinoma. J Clin Oncol. 2016;34(14):1660–1668. doi:10.1200/JCO.2015.64.880826951309
- Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5(9):769–784. doi:10.1038/nrd213316955068
- Zibelman M, Wong YN, Devarajan K, et al. Phase I study of the mTOR inhibitor ridaforolimus and the HDAC inhibitor vorinostat in advanced renal cell carcinoma and other solid tumors. Invest New Drugs. 2015;33(5):1040–1047. doi:10.1007/s10637-015-0261-326091915
- Wood A, George S, Adra N, Chintala S, Damayanti N, Pili R. Phase I study of the mTOR inhibitor everolimus in combination with the histone deacetylase inhibitor panobinostat in patients with advanced clear cell renal cell carcinoma. Invest New Drugs. 2020;38(4):1108–1116. doi:10.1007/s10637-019-00864-731654285
- Yang YP, Hu LF, Zheng HF, et al. Application and interpretation of current autophagy inhibitors and activators. Acta Pharmacol Sin. 2013;34(5):625–635. doi:10.1038/aps.2013.523524572
- Haas NB, Appleman LJ, Stein M, et al. Autophagy inhibition to augment mTOR inhibition: a phase I/II trial of everolimus and hydroxychloroquine in patients with previously treated renal cell carcinoma. Clin Cancer Res. 2019;25(7):2080–2087. doi:10.1158/1078-0432.CCR-18-220430635337