Abstract
Over 50 years of cancer therapy history reveals complete clinical responses (CRs) from remarkably divergent forms of therapies (eg, chemotherapy, radiotherapy, surgery, vaccines, autologous cell transfers, cytokines, monoclonal antibodies) for advanced solid malignancies occur with an approximately similar frequency of 5%–10%. This has remained frustratingly almost static. However, CRs usually underpin strong durable 5-year patient survival. How can this apparent paradox be explained? Over some 20 years, realization that (1) chronic inflammation is intricately associated with cancer, and (2) the immune system is delicately balanced between responsiveness and tolerance of cancer, provides a greatly significant insight into ways cancer might be more effectively treated. In this review, divergent aspects from the largely segmented literature and recent conferences are drawn together to provide observations revealing some emerging reasoning, in terms of “final common pathways” of cancer cell damage, immune stimulation, and auto-vaccination events, ultimately leading to cancer cell destruction. Created from this is a unifying overarching concept to explain why multiple approaches to cancer therapy can provide complete responses at almost equivalent rates. This “missing” aspect provides a reasoned explanation for what has, and is being, increasingly reported in the mainstream literature – that inflammatory and immune responses appear intricately associated with, if not causative of, complete responses induced by divergent forms of cancer therapy. Curiously, whether by chemotherapy, radiation, surgery, or other means, therapy-induced cell injury results, leaving inflammation and immune system stimulation as a final common denominator across all of these mechanisms of cancer therapy. This aspect has been somewhat obscured and has been “lost in translation” to date.
Introduction
The low clinical efficacy of therapies for advanced cancer, despite over 40 years of intensive funding and research, has been recently noted and commented upon by many government authorities and scientific and funding bodies.Citation1–Citation4 This interest is principally because of the continually escalating costs associated with clinical cancer care, with an aging population in Western countries, and the slow progress in curing cancer.Citation1–Citation4 Patients with most major advanced cancers can expect a complete response (CR) rate, where all cancer disappears, of about 5%–10%, and this has remained frustratingly almost static. However, CRs typically underpin durable 5-year survival.Citation5–Citation7
In broad terms, current paradigms in cancer therapy revolve around the capacity of chemotherapeutic agents to interfere with cancer cell division, signaling, or cancer cell intracellular metabolism, using chemotherapy, radiotherapy, surgery, vaccines, autologous cell transfers, cytokines, monoclonal antibodies, and other modalities to cause direct cancer cell killing. An apparent divergence in this concept has been the antiangiogenic agents, which interrupt tumor vasculature and blood supply. However, it has been noted for over 100 years that immunological manipulations alone can similarly produce some remarkable regressions of cancer, apparently quite separately from the standard presumed mechanisms of cell death from cytotoxic and antiangiogenic agents. Evidence for this derives from reports of serendipitous infection or infective inoculation of cancer patients, especially after surgery, from the use of vaccine preparations, including bacillus Calmette-Guerin and bacterial extracts; or more recently, through cytokines, such as recombinant interleukin (IL)-2, and monoclonal antibodies such as the anti-cytotoxic T-lymphocyte antigen (anti-CTLA)-4 agents, which all possess the capacity to induce cancer regression in a clinically indistinguishable manner to that produced by standard cytotoxic modalities.
Cytotoxic chemotherapies, radiation therapy, surgical resection, vaccine therapies, modified viral agents, laser therapy, cryotherapy, radiofrequency ablation, infectious agent byproducts, electroporation of antigens, cytokine therapies (eg, IL-2, IL-12, interferon [IFN]-α, IFN-γ, tumor necrosis factor [TNF]-α), dendritic cell therapies, autologous cell therapies, natural agents (eg, curcumin, vitamin A, vitamin C), and monoclonal antibody therapies are all capable of generating a small number of random/unpredictable CRs, and when these occur they are frequently associated with long-term durable survival.
A paradox seems to exist, because many divergent modalities achieve a remarkably similar 5%–10% CR rate. How can this be?
Many agents with apparently very widely divergent and diverse biological mechanisms of action are capable of causing a limited number of random CRs, partial responses, and even stability of disease. By drawing observations together, it can be reasoned that there is a distinct possibility that a “final common pathway” must exist to produce these unpredictable CRs. This “final common pathway or mechanism” might likely be operational to collectively explain these observations, regardless of the duration and type of the initiating therapy.
The realization and consolidation over the last 20 years that (1) chronic inflammation is intricately associated with cancer, and (2) that there is a delicate constant immune system dynamic balance between responsiveness on one hand and tolerance of cancer on the other, provides a greatly significant insight into the ways cancer might be more effectively and predictably treated.
This review provides some observations from the literature and recent conferences and proposes some emerging reasoning capable of explaining these phenomena in terms of a final common pathway(s) of immune stimulation and “auto-vaccination events” ultimately leading to cancer cell destruction that can produce the CRs and which affect patient survival.
Current paradigms are explored relating to mechanism of actions of current therapeutic approaches to cancer, some important questions are raised, and some considerations are proposed that may reasonably offer some unifying explanation for these seemingly disparate observations.
Cancer therapies and mechanisms of action
Standard chemotherapy approaches
Standard cytotoxic chemotherapy operates on the well-accepted and entrenched paradigm of cytotoxicity towards cancer cells during mitotic division and active metabolism. Mechanisms for cellular toxicity define different classes of agent to include alkylation of DNA (eg, cyclophosphamide, chlorambucil, cisplatin, carboplatin, oxaliplatin, melphalan, dacarbazine), anti-metabolites (eg, methotrexate, azathioprine, mercaptopurine), microtubule spindle poisons (plant alkaloids and terpenoids, eg, vinca alkaloids, vincristine, vinblastine; podophyllotoxin; taxanes, including docetaxel and paclitaxel), topoisomerase inhibitors (eg, type 1 include irinotecan and topotecan, and type 2 inhibitors including amsacrine, etoposide, etoposide phosphate, and teniposide). Cytotoxic antibiotics or antineoplastics (eg, dactinomycin [or actinomycin-D], doxorubicin, epirubicin, bleomycin, mitomycin-C).Citation5
In many solid cancers, at any one time-point, the proportion of actively dividing cancer cells is estimated to be of the order of 20%–40% of the total cell population within the tumor mass(es).Citation5 This is because the cells in a malignant mass divide asynchronously and haphazardly. This fact means that with any one dosing administration the maximum proportion of cancer cells that can be influenced by the cytotoxic agent(s) is around 30%, leaving some 70% unable to be effected by the agent(s). Repeated dosing is assumed to randomly influence the remaining cancer cells in the tumor mass when those cells commence divisions at later time-points. When a particular cancer cell, or clone of cancer cells, can be influenced by the cytotoxic agent is currently unpredictable and is determined by probability – defined by a series of mathematical parameters, including the timing frequency of the agent’s delivery, the half-life of the agent(s), superimposed upon the frequency of cell division within the cancer mass(es), the proportion of cancer cells actively dividing, and the extent of cancer cell killing with each dosing. Vascular injury, especially endothelial damage, may compound these parameters and enhance cellular killing.
It is also well recognized that cytotoxic agents not only affect dividing cancer cells but also kill dividing normal cells in a range of tissues, most notably rapidly dividing normal cell populations. Nonmalignant cells typically divide in a synchronous fashion during clonal expansion to either repopulate tissues for replacement of naturally dying or injured cells, or in the case of the immune system, in response to stimulation from other signals, typically the presence of “nonnormal” cellular antigen or inflammatory responses of other types; for example, traumatic cellular injury. Rapidly dividing normal cells under normal circumstances of clonal expansion are particularly vulnerable to injury from exposure to cytotoxic agents in the course of cancer therapy. Cells in the bone marrow, digestive tract, and hair follicles are often affected, directly causing a range of well recognized clinical effects such as myelosuppression (both innate and adaptive immunosuppression, detected as neutropenia and lymphopenia respectively), susceptibility to infection, gastritis and diarrhea, mucositis, perforation, and alopecia. Because normal, rapidly dividing cells are exquisitely sensitive to cytotoxic agents, the consequent side-effects of cytotoxic therapy are not uncommonly significant in producing dose limitation and cessation, causing either delayed therapy, dose reduction, poor compliance, alteration of therapy type, complete cessation of therapy, or rarely, early death. Exploration of the effects of cytotoxic agents on the bone marrow and immune system has yielded some observations that are relevant to understanding how chemotherapies may actually work. The effects of cytotoxic agents on bone marrow are to remove any clonally expanding subpopulations of immunological cells. The innate immune response cells are particularly sensitive to cytotoxic agents, leading to reduced neutrophil and monocyte counts in the circulation and tissues. Furthermore, the subpopulations of lymphocytes, both effector and regulatory arms, including both CD4 and CD8 cells, are also highly sensitive to the effects of cytotoxic agents. Which cell subpopulations are preferentially killed or affected is largely determined by which subpopulations of cells are actively/synchronously and discretely dividing and expanding at the time of administration of the chemotherapeutic agent. The precise nature of the immune system micromanipulation that occurs with any one chemotherapy dose is therefore largely a consequence of the time that the dose is delivered into the patient, the dose size, and the half-life of the agent (which determines the duration of effect of the agent). There is emerging clinical evidence that the immune system undergoes dynamic regular homeostatic oscillations in patients with advanced cancer, and therefore there is likely a regular alternating expansion of the effector arm, followed by the regulatory arm, of the immune system that occurs in most, if not all, cancer patients.Citation6–Citation9 This means that the timing of administration of the chemotherapeutic agent within this dynamic framework is also likely to be critical in determining not only the side-effects and morbidity, but in determining the direction that the immune system is driven – either activation/responsiveness or inhibition/tolerance, and thereby determining the clinical outcome. This would explain a range of common clinical observations that clinicians typically see and that are reported in the literature and many studies. These include, the random and unpredictable nature of many clinical responses, the heterogeneity of observed responses, the development of a successful response after previous poor responses from repeated dosing, and the variable incidence of side-effects, such as neutropenia or lymphopenia (even in the same patient with sequential dosing).
Importantly, in addition to cell killing, which is assumed to be the main action of most cytotoxic agents, cell injury also occurs. Both cell killing and cell injury are associated with “danger signals” issued to the immune system, both innate and adaptive, which are known to be powerful stimuli and drivers of immune reactivity. Therefore, both antigen release and danger signals, the two most powerful natural drivers of the immune response in evolutionary terms, form an integral part of any type of cell damage, lethal or otherwise, induced by chemotherapeutic or other means.Citation10
The predictable translation from the often-successful mouse experiments to the human situation has been problematic and disappointing. The sequence of events for successful tumor cell inoculation, and the timing of therapy, has not yet been accurately addressed. There is a paucity of careful time-course studies published that collect serial/daily blood data over 2–3 weeks or more. Consequently, the accurate experimental description of the immune kinetics following tumor cell inoculation and the initiation of the antitumor immune response, and its subsequent attenuation or regulation over time, has not been fully elucidated. Without this extended serial data, the finer details of the “bimodality” issues that precisely influence cancer therapies of most types, including chemotherapy and cytokines, cannot be fully observed, appreciated or understood, in either animal or human studies.
Radiation therapy approaches
Radiation therapeutic approaches utilize free-radical producing effects of ionizing radiation to produce DNA damage and consequent cell death.Citation5 High-energy X-rays gamma-ray emitting sources, particles, and electrons are typically used for radiation therapy. External beam radiation therapy is the conventional form of externally delivered focused X-rays (photons) to a localized tumor deposit, often from two separate directions to reach the same area. Stereotactic radiation (radiosurgery) uses focused external beam radiation therapy to target a well defined tumor with a high degree of precision using very fine, detailed three-dimensional imaging scans with coordinates to plot the area more specifically. It is increasingly being used, for example, with lesions deep within the brain. Three-dimensional conformal radiation therapy is an approach that better permits radiation delivery to conform or fit the shape of the tumor, reducing the relative toxicity to surrounding normal tissues. Intensity-modulated radiation therapy is a modification of this, with improved ability to conform the treatment volume to concave tumor shapes, like spinal cord or internal organs against bone. Brachytherapy is used to place or implant, temporarily or permanently, the radiation emitting source(s) close to the cancer mass to release usually, either beta-particles or gamma-rays (depending on the source chosen) to cause local cancer cell killing. Systemic radioisotope therapy is where chemical properties of the isotope itself, such as radioiodine, or when chemically linked with another molecule (eg, hormone or monoclonal antibody) are capable of more selectively delivering radioisotopes, usually via ingestion or the bloodstream, to a specific organ or tumor site for more selective action. Examples are: anti-CD20 monoclonal antibody conjugated to yttrium-90, metaiodobenzylguanidine (MIBG) to treat neuroblastoma, oral iodine-131 for thyroid cancer, and yttrium-90 for neuroendocrine tumors. Radioactive microspheres can be used to inject selectively into a tumor circulation to radioembolize liver metastases, for example. Particle therapy is a form of delivery of high-energy radioactive particles such as protons, neutrons, or positive ions to cancer tissues. Electron (beam) therapy is generally considered separately, but also imparts radiation energy, although with less penetration, and is used for more superficial tumors like skin cancer masses and breast cancers.Citation5
The common effect of all of the above forms of radiation therapy is delivery of high-energy radiation damage to cancer cells to cause cellular DNA damage and either activation of programmed cell death (apoptosis) or necrosis or cell injury, leading to danger signals and antigen release.Citation10 In either event, inflammation occurs and immune system activation/regulation results. Injury to normal dividing cells within the path of the radiotherapy beam or from systemic administration is not uncommon and results in a range of commonly observed side-effects of therapy. These include damage to epithelial surfaces such as desquamation, burn injury, erythema, swelling, local tenderness, ulceration, mucosal loss and bleeding, oral ulcers (mucositis), intestinal problems such as diarrhea and nausea, and immunosuppression. Later effects include fibrosis, vascular angiomas, strictures, and malignancy.
The radiation is delivered either as a single dose, or more commonly, for external forms of radiotherapy, as multiple divided fractions over time. With respect to the immune system, the intravascular cells circulate through the blood vessels in and around the cancer mass(es) and enter the cancer interstitium and the surrounding normal tissues, including within adjacent lymphoid and bone marrow tissues. These leukocytes entering the region are exposed to the direct effects of radiation, and the consequent effects of free radical formation. In situations where the body region(s) exposed to radiation are extensive or if therapy is prolonged, lymphoid tissues achieve greater exposure, and the effects on the immune system can be appreciable, evidenced by increased infection rates. Necrosis caused by radiotherapy can induce inflammation at the site(s) of radiation therapy, leading to exposure of relatively more immunological cells attracted to the area(s), which at the time of exposure, may be actively dividing and vulnerable.
Radiation damage to immunological cells can cause cell death from apoptosis, and the precise immunological cell population influenced will depend on which arm of the immune equation (effector or inhibitory) is actively expanding/dividing at the time of exposure of the radiotherapy dose and its toxic byproducts. Therefore, the effects of each radiation dose will likely be pivotal upon which arm of the immune system is most active at the time of the administration. During radiation exposure, if the regulatory arm is most active, that will be ablated; alternatively, if the effector arm is most active, that will be injured. The balance of the extent of the ablation with respect to the overall total immune system capacity will therefore largely determine the overall damage to the immune system and the precise functional effect of each radiation dose or of repetitive effects caused by repeated dosing. The effects of radiation on the regional and systemic immune system is, in principle, analogous to the effects of chemotherapy, although potentially not as extensive due to the beam exposure, unless large marrow areas are included in the radiation field(s). Moreover, radiation damage to cancer cells can induce antigen release and can induce a direct anticancer immune response capable of driving either activation or tolerance.Citation11–Citation14
Surgical approaches
Surgery is widely assumed to remove cancer by resecting the main tumor mass(es), thereby curing the patient. If no cancer cells remain, this explanation might be readily acceptable in biological terms; however, the literature is increasingly recording studies that show that cancer cells circulate intravascularly widely within the body away from the primary tumor site, even in early stage cancers. This phenomenon has been observed for multiple tumor types, both before and after surgery, and is not always clearly associated with an adverse prognosis or a more advanced stage of the cancer. Moreover, many instances are well-recognized anecdotally by practicing surgeons where the cancer is macroscopically removed, but residual cancer cells are reported at the margins by the pathologist, however in an appreciable number of these cases the tumor never regrows at that site. Moreover, in reports of resection of multiple metastases, for a range of cancers, long-term cure is possible, despite clearly widespread hematogenous dissemination of the cancer to multiple body sites.Citation15–Citation21 However, the extent of patient survival after surgery is highly influenced by disease status and patient selection, and is consequently variable. How can these observations be explained? Surgery usually aims to remove the cancer in its entirety, or at least as a near-entire debulking procedure to remove the main cancer mass(es) prior to other therapy. It is also well-recognized that surgery is inherently traumatic to normal and cancer tissues; indeed, often the aim is to not directly visualize cancer tissue at all during surgical extirpation while resecting a suitable margin of normal surrounding tissue(s). Such local tissue trauma is usually quite appreciable to anyone who has had surgery, and biologically, causes a significant and measurable cascade of cytokine release and immune stimulation to occur. Immunosuppression after major open surgery is well documented, and usually lasts 7–14 days after surgery, unless infection supervenes.Citation5 In general terms, during this period, pro-inflammatory cytokines are transiently suppressed, and pro-regulatory/inhibitory cytokines are elevated. The immune system stimulation that occurs after surgery is usually thought of as nonspecific; however, it now appears that stimulation of preexisting immune responses against the cancer (and other preexisting inflammatory states) may well occur to effectively “augment” these ongoing immune responses. This boosting of the underlying native preexisting immune response(s), may explain why some cases of overtly residual cancer after surgery never re-grow to produce a “cancer recurrence”. Moreover, cancer antigens are released during surgery, especially if the cancer tissue is breached or traumatized in some manner, even through ischemia. Such an antigen release would effectively constitute an “auto-vaccination” event, with capacity to stimulate the immune system in an immediate and prolonged way. The direction that the immune response is driven would potentially be dependent upon the extent of the stimulation from trauma (necrosis and danger signals) and the direction that the immune response is pointed – responsiveness or tolerance – at the time of surgery.Citation10 This explanation is both plausible and supported by some evidence, and if shown to be true, then virtually every cancer surgical procedure would potentially constitute such a significant “auto-vaccination” event, many of which are clinically successful.
Hormonal inhibitors
Initial discoveries that some breast cancers were hormonally dependent and that hormonal withdrawal using oophorectomy could halt metastatic breast cancer growth for premenopausal women were instrumental. Initial observations that administration of estrogen or progesterone to some women with certain breast cancers could stabilize disease progression or induce complete cancer regression, while in others could promote progression, was pivotal for the ultimate development of a range of new agents and approaches to therapy for hormone sensitive cancers. The discovery that blockade of hormone receptors on certain cancer cells could modulate growth patterns of these cells both in vitro and in vivo provided a massive step forward in so-called hormone sensitive cancers. Tamoxifen was the first of the effective specific agents, initially thought to competitively blockade the estrogen receptor (ER) on breast cancer cells. It quickly became apparent that not all cancer cells within a cancer mass expressed ER, that the intensity of expression varied amongst those cells that did, and that some breast cancer cells were apparently responsive even if the ER was undetectable in the patient’s cells.Citation5 So that heterogenous ER patterns of expression within and between tumors was common and that although this helped to determine responsiveness it was not always clinically predictive. The progesterone receptor (PR) expression also appeared to play a role, but although this was important, it was not as pivotal as ER. Further, aromatase inhibition, using dinitrile (cyanide-like) agents such as anastrozole, exemestane, and letrozole, appeared to provide a more complete mode of deprivation of sex hormone production through blockade of adrenal steroid hormone synthesis, thus removing the actions of estrogen and progesterone through the ER and PR on breast cancer cells. Another approach, used for premenopausal women with breast cancer, is the use of luteinizing hormone blockers such as goserelin (Zoladex®; AstraZeneca, London, UK) to prevent ovarian production of sex steroids, analogous to surgical oophorectomy.Citation5
Prostate cancer cells are dependent on testosterone for adequate growth in many situations and express the androgen receptor (AR) heterogeneously within a prostate cancer, and with variable intensity, in a similar manner to that seen with breast cancer and ER/PR expression. Therefore, removal of testosterone stimulation of AR is used for prostate cancer therapy to reduce growth rates and in some cases causes complete regression.Citation5
The basic methods of androgen deprivation are castration, surgical (orchidectomy) or biochemical gonadal testosterone depletion (luteinizing hormone-releasing hormone [LHRG] agonists, and LHRH antagonists). The extent of testosterone blockade from biochemical agents compared with healthy males can vary but is often 90%–95%. Estrogen was used widely initially to treat prostate cancer, however synthetic diethylstilbestrol was associated with serious cardiovascular problems. The net effect of estrogen is to block testosterone production by the testes by blocking hypothalamic gonado-trophic hormone release. Anti-androgens produce androgen receptor blockade, including within the prostate cancer to reduce growth. Combined androgen blockade utilizes both castration and anti-androgen agents to ablate testosterone and its effects on prostate cancer cells.Citation5
Ovarian cancer cells appear to respond to estrogen and progesterone blockade, using tamoxifen and aromatase inhibitors in some instances, in a similar fashion to breast cancer. This avenue is being actively explored.
The net effects of hormonal blockade are deprivation of essential growth signals for cellular survival and expansion. Without those essential hormonal influences, the cells become quiescent, are damaged, or die. Death is usually by apoptosis, but occasionally necrosis occurs. In either case, antigenic exposure is produced and the immune system is exposed to this tumor antigenic stimulation. The net outcome of the exposure to tumor antigen would be expected to be determined by whether the immune system was displaying the predominant direction of either responsiveness or tolerance at the time.
Anti-angiogenic agents and vascular inhibitors
Anti-angiogenic molecules act by inhibiting formation of endothelium via a variety of approaches. Vascular endothelial growth factor (VEGF) inhibitors, such as bevacizumab (Avastin ®; Genentech, South San Francisco, CA), are monoclonal antibodies which bind to VEGF to prevent activation of the VEGF receptor. Other angiogenesis inhibitors, eg, sorafenib and sunitinib, bind to surface endothelial cell receptors or to downstream signaling pathway proteins to block angiogenesis. Bevacizumab has been used for therapy of metastatic colorectal cancer, some non-small cell lung cancers, metastatic renal cell cancer, and glioblastoma.Citation5 It was recently withdrawn from approval for use in advanced breast cancer due to evaluated inefficacy. Anti-angiogenic agents include sorafenib (Nexavar®, Bayer, Leverkusen, Germany), approved for hepatocellular carcinoma and kidney cancer, sunitinib and everolimus for both renal cancer and neuroendocrine tumors, and pazopanib for renal cancer.
Anti-angiogenic agents prevent new vascular formation and/or disrupt vascular integrity, and thereby reduce nutrition to the tumor, which can produce effective cancer cell killing. They may be combined sequentially or concurrently with other chemotherapeutic agents. The net result is cancer cell death by either necrosis or apoptosis, leading to antigenic release with immune system priming with tumor antigens and danger signal release.Citation10 Together, these new agents have provided modest and inconsistent improvements in overall survival. The effects on the immune system are less clear and include injury to lymphoid tissues, including lymph nodes, spleen, and bone marrow. The timing of the antigenic stimulation of the immune system pathways is potentially important, as the direction that the immune system will be driven – responsiveness or tolerance – might reasonably be expected to depend upon the relative effector versus regulatory balance that exists at the time of stimulation.
Metabolic pathway inhibitors and monoclonal antibodies
HER-2 (human epidermal growth factor receptor-2; proto-oncogene Neu, receptor tyrosine-protein kinase erbB-2, CD340, or p185) is a surface-bound cell membrane receptor tyrosine kinase enzyme encoded by the ERBB-2 human gene, with overexpression correlated with higher breast cancer aggressiveness in growth and increased disease recurrence. HER-2 is normally involved in the signal transduction pathways leading to cell growth and differentiation, but in about 30% of breast cancers amplification of the HER-2/neu gene or overexpression of its protein product occurs.Citation22–Citation25 Overexpression of HER-2 also occurs in other cancers such as ovarian, gastric, esophageal, and uterine (serous endometrial) carcinomas. Trastuzumab (Herceptin®; Genentech) is a humanized murine monoclonal antibody directed to one part of the HER-2 receptor, and its identified mechanisms of action are suppression of angiogenesis, cell cycle arrest during the G1 phase (producing reduced proliferation and cell death), and induction of cell killing by immune cells through antibody-dependent cell-mediated cytotoxicity.Citation25
B-Raf is a member of the Raf kinase family of serine/threonine-specific protein kinases and is a critical enzyme protein for regulation of the mitogen-activated protein kinase (MAPK)/extracellular signal-related kinase (ERK) signaling pathway, important for cell division, differentiation, and secretory function.Citation26 BRAF gene mutations can be inherited or arise later as an acquired oncogene. Malignant melanoma, non-Hodgkin lymphoma, colorectal cancer, papillary thyroid carcinoma, colorectal cancer, hairy cell leukemia, non-small cell lung carcinoma, and lung adenocarcinoma are associated to variable degrees of different mutations (notably the V600E variant).Citation27–Citation31 Over 25 different variant mutations are described. B-Raf inhibitors have been described and used clinically in trials for therapy for melanoma and other cancers overexpressing specific mutations. Examples are PLX4032 (RG7204; Plexxikon/Hoffmann–La Roche; vemurafenib), and GSK2118436 and GSK1120212, and some more general B-raf inhibitors including GDC-0879, PLX-4720, and sorafenib tosylate. The mechanism of action is thought to be by binding to the V600E mutant form of the B-Raf enzyme protein inducing programmed cell death. Necrosis of tumor masses has been suggested, and associated antigen and danger signal release would be likely.Citation10 There may be a paradoxical stimulation of growth through wild-type nonmutant forms of B-Raf. Off-target side effects include induction of skin cancers. The overall response rate with vemurafenib was 46% and the CR rate was 6%. When comparing with non-B-raf selected therapies, these rates should logically be halved (ORR 23%, CR 3%).
MEK inhibitors have gained recent interest for cancer therapy. MAPKs are serine/threonine-specific protein kinase enzymes that catalyze a cascade of intracellular enzymes, the MAPK/ERK pathway, in response to a wide range of extracellular stimuli (cellular stress, including osmotic stress, heat shock, and pro-inflammatory cytokines and mitogens) for a broad range of cellular functions; for example, gene expression, division and expansion, differentiation, proliferation, and cell survival/apoptosis.Citation32 When activated, Ras activates RAF kinase,Citation29 which phosphorylates and activates MEK (MEK1 and MEK2). MEK activates a MAPK. RAF, MEK, and MAPK are all serine/threonine-selective protein kinases. Some MEK inhibitors include XL518, CI-1040, PD035901, selumetinib, and GSK1120212. MEK inhibitors are currently being trialed in combination with B-Raf inhibitors.
The precise mechanisms of action of each inhibitor are unclear, despite the site of proposed blockade being fundamentally physicochemically understood. The reason for this is that the effects upon many groups of normal cells in vivo have been incompletely studied to date, including those of the immune system. All human cells contain B-Raf, C-Raf, MEK, and EGF, usually in wild-type form, although it is increasingly being appreciated that mutant forms of all of these enzymes are far more widespread than initially thought. This means that normal cells are being influenced by these agents, which are principally “targeted” at the cancer cells, but are accidentally influencing normal cell function in the course of cancer therapy, accounting for many of the observed and reported “off-target” side-effects of these agents in vivo.
Moreover, the action of these agents on cancer cells is to slow growth (although occasionally activation is reported), and to cause cancer cell death, with consequent regression of cancer masses in some cases. Cell death induces antigenic release, immune reactivity, danger signals, and an immune response, often with consequent inflammation.Citation10 The direction and magnitude of this immune response is dependent upon the extent of antigen release, the antigenicity of this, and likely the balance and direction of the immune response at the time of stimulation.
Immunological agents
Cytokines
Cytokines of various types have been used for cancer therapy for a number of years and in various combinations with other agents. IL-2, initially known as T-cell growth factor, has perhaps been the most prominent and sustained agent in use for about two decades and still remains in wide use. A recent review demonstrated data from multiple studies concerning melanoma and renal cell carcinoma and revealed an overall CR rate of about 7.6% (from further analysis of the reported summarized data).Citation32 IL-2 binds to a trimeric receptor complex, which includes the IL-2 receptor alpha (CD25), IL-2 receptor beta (CD122), and a common gamma chain (γc), which is shared by all members of the family of cytokines including IL-4, IL-7, IL-9, IL-15, and IL-21. IL-2 receptor (IL2R) complex binding activates the Ras/MAPK, JAK/Stat and PI 3-kinase/Akt signaling modules in T-cells and several other types of cells. Importantly, and a point often overlooked, is that many cytokine receptors, like for example IL2R are expressed transiently on T-cells over a duration of about 6–8 hours and are then lost from the T-cell surface.Citation33–Citation48 It is only during IL2R expression that IL-2 can activate the T-cell to induce cell division and clonal expansion of T-cells. However, IL2R is expressed on both T-effector and T-regulatory cells, and current evidence shows that the immune system oscillates in the intensity of the inflammatory response in most, if not all, patients with advanced cancers. This raises the distinct likelihood that the immune system fluctuates in the sequential repetitive cellular expansion causing alternate homeostatic activation and then inhibition. If this proves indeed to be the case, then T-cells from either the T-effector arm or T-regulatory arm of the immune response would express and then lose IL2R on their surfaces. During transient IL2R expression, either T-effector or T-regulatory cells would alternately become responsive to IL-2 in the tumor microenvironment. The “bimodal” and transient nature of IL2R expression would explain the many apparently conflicting observations in the literature for over three decades or more, where IL-2 appeared to stimulate either a predominantly effector or predominantly regulatory immune response, inducing either detectable responsiveness or tolerance. Hence, the direction that the immune response is driven – overall responsiveness or tolerance – will depend on when the IL2R is being expressed, on which cells, and whether IL-2 cytokine is present.
Many other cytokines and their receptors are becoming increasingly recognized as possessing “bimodal” and transient actions on both T-effector and T-regulatory cell populations, capable of inducing either responsiveness or tolerance. These now include IFN-γ, IFN-α, TNF-α, antigens of many types, IL-17, IL-12, and recently IL-10.Citation38–Citation46 Consequently, these “bimodal” attributes are often reported as being “paradoxical,” probably because the time domain has not yet been accurately mapped using daily/near daily data over an extended period (10–21 days or more) to resolve the opposing actions over time. Using combined IL-2 and CTLA-4, an increased CR rate of 17% with strong durable survivals has recently been reported by Rosenberg et al.Citation48
CTLA-4 inhibitors
CTLA-4 (cytotoxic T-lymphocyte antigen 4; CD152) is a protein that is a member of the immunoglobulin superfamily, which is expressed on the surface of T-effector and T-regulatory cells and transmits an inhibitory signal to T cells for downregulation of immune responses. CTLA-4 and the T-cell costimulatory protein CD28 both bind to CD80 and CD86 (also called B7) on antigen-presenting cells. CTLA-4 induces an inhibitory signal to T cells, whereas CD28 provides a stimulatory signal.Citation48–Citation50 T-cell activation via the T-cell receptor and CD28 leads to increased CTLA-4 expression as an inhibitory feedback loop for B7 molecules (ie, CD80 and CD86). CTLA-4 gene mutations and polymorphisms are associated with insulin-dependent diabetes mellitus, Graves’ disease, Hashimoto’s thyroiditis, celiac disease, systemic lupus erythematosus, primary biliary cirrhosis, multiple sclerosis, and other autoimmune diseases. CTLA-4 blocking monoclonal antibodies (eg, ipilimumab [MDX-010; Yervoy] and tremelimumab), act by binding to parts of the CTLA-4 molecule, thereby blocking the inhibitory signal from CTLA-4 to T-cells, and have been used for therapy of malignant melanoma, prostate cancer, and other cancers. Overdrive of the immune response with induction of autoimmunity can occur, sometimes causing severe adverse events, including treatment-associated death. The CR rate for patients with advanced melanoma is 1.5%, with an overall response rate of 7% and prolongation of survival of about 10 months before death. As with the IL-2/IL2R circuitry, CTLA-4 has “bimodal” expression/activity on effector and regulatory T-cell populations.Citation46,Citation47 Recently Qureshi et al has suggested CTLA-4 exerts a similar mechanism of expansion control on T-effs and T-regs.Citation51 Consequently antibodies (with half-lives of 2–3 weeks) targeting CTLA-4 could indiscriminately blockade either or both T-eff and T-reg populations accounting for the unpredictable/random and low number of clinical responses seen to date, and explaining the autoimmune side-effects.
Vaccines
Many vaccine types have been trialed against established nonresectable cancers in humans, with variable and limited success. However, CRs are well reported in many studies, ranging from 2.0%–18.9%.Citation8,Citation48,Citation52–Citation55 Recent interest over the last 15 years or so has focused on approaches using dendritic cells to improve antigen presentation to the immune system both systemically and locally within the tumor microenvironment. This appeared initially attractive during a time when it was strongly held that the problem with anticancer immune responses lay significantly with failure of adequate antigen presentation to the immune system. Combined accrued results from 51 dendritic cell (DC) vaccine studies in almost 1000 patients were reviewed recently and indicated that the CR rates for multiple different DC approaches were between 2.3% and 3.5%.Citation56 A recent report presents a new insight into antigen stimulation by demonstrating that frequent repetitive dosing of vaccine and the persistence over a long duration of 6 months or more, despite the absence of initially apparent clinical effects, may provide important clues towards improving clinical efficacy.Citation8 This and other reports in the literature strongly indicate that antigen presentation per se may not be the problem, as the immune system in the cancer patient appears to be adequately detecting tumor antigen, but is being effectively downregulated to prevent reactivity, resulting in tumor tolerance. The fact that CRs can be obtained from vaccination alone against cancer (as from cytokines and CTLA-4 MoAb’s used alone) shows that direct cancer cell killing using cytotoxic agents is not necessarily required to achieve strong, durable CRs.
Conclusion
Strong durable CRs in human cancers of most types are able to be derived by surprisingly diverse presumed and described mechanisms of action, and are observed to occur spontaneously as well at a low rate without any overt apparent intervention. The effectiveness of the cancer research effort has been seriously criticized lately, indicating that small gains in disease-free progression of the order of 10 days to 3 months are unacceptable, and that “It is just not true . . . how successful the cancer research enterprise is.”Citation4,Citation57 Research approaches have evolved into segmented, isolated disconnected efforts in highly specific areas of cancer investigation. The individual proponents of these therapeutic approaches have put forward and propounded individual mechanisms of action; for example, actions relying on direct cellular cytotoxicity (chemotherapy and radiotherapy), metabolic disjunction or suppression (pathway inhibitors), resection (surgery), or immunological manipulation (cytokines, CTLA-4 MoAbs, vaccines). The observed rates of CR across almost all of these approaches of anticancer therapy for advanced human cancers (excepting testicular cancer, acute childhood leukemia, choriocarcinoma, and perhaps some lymphomas) are of the order of 0%–20% by most methods, averaging at around 5%–10% overall. Moreover, many approaches regarded as current “standard-of-care” therapy have CR rates of well less than 10% (eg, lung, melanoma, mesothelioma, pancreatic cancers) and very low 5-year survival rates.
Two perhaps surprising points are evident from this broad clinical observation: (1) that the similarly uniformly low rates of CR are obtained despite a wide and diverse range of modalities being used for cancer therapy, and (2) that purely immunological therapies can produce CRs at approximately the same rates as ablative therapies. These points were raised and discussed at several levels at the American Society of Clinical Oncology in Chicago, in June 2011, at the recent Society of Immunotherapy of Cancer Conference in Washington DC, November 2011, the NCI satellite symposium in November 2011, and also at the Australasian Society of Immunology meeting in Adelaide, in December 2011.
How can these observations be logically explained? The observation that all of the available anticancer therapies, apparently regardless of their mechanism of action and combination, appear to arrive at a relatively common low rate of CR and that this has remained essentially unchanged for over 40 years, profoundly suggests that a common final biologically limiting pathway may be present to account for the above findings. Such a common mechanism for cancer control would be an important point to appreciate, and would explain our relative lack of progress in understanding how to better advance the efficacy of cancer therapy to date. That is, to appreciably increase CRs and survival, rather than to aim for increasingly marginal improvements with complicated and often expensive approaches. Increasing the CR rate should be our main goal as espoused recently by Tuma.Citation57
What would this common overarching pathway then be? It is instructive that for each of the myriad forms of cancer therapy – chemotherapy, radiotherapy, surgery, and others – there are cancer cells injured or killed as the main byproduct of the mechanisms of action. The immune system – both innate and adaptive – is designed through evolution to detect aberration of the cell membrane. Magnetic resonance spectroscopy studies have demonstrated detectable differences in the composition of the cell membrane as the cell evolves from normal towards malignancy.Citation58,Citation59 These sometimes subtle changes, notably in glycolypids, rather than the peptide component where our main focus has typically resided, are capable of inducing antigenic profile alterations in cells, which the immune system is then capable of detecting and reacting towards. Cell damage, both cancer and normal, is thereby an important and powerful stimulus for tissue inflammatory responses – initially innate, then adaptive – with both arms of immunity progressing simultaneously after cell danger signals have been liberated.Citation5,Citation8,Citation10 The immune system is initially stimulated/initiated, and then terminated, as a normal homeostatic part of the (acute and chronic) inflammatory response, in consort with simultaneous hematological, endocrine, and sometimes neural responses, as part of the tissue trauma or injury response. Pro-inflammatory cytokines and chemokines (which can also be immune suppressive/regulatory) are released upon tissue injury, and are more likely to be cancer-directed upon cancer cell injury and antigen exposure. With evidence that the immune system is already detecting the presence of the cancer, as it does with any form of cellular or tissue aberration, through aberrant glycolipid and other surface markers described during (even early) malignant cellular transformation, Citation58,Citation59 it is clear that the immune system must be responding, even if in a (subclinical) attenuated manner, to signals from (nonnormal) cancer cells. For pure immunostimulant agents, such as IL-2, IL-12, or CTLA-4 blocking antibodies, to have any effect at all in producing CRs, a preexisting, ongoing, immune response against the cancer, with existing presentation of tumor antigens, must have been present but in a downregulated state. This is supported by knowledge that the ineffectiveness of the immune response in patients with advanced cancer has been increasingly recognized to be associated intimately with the presence of a predominance of local and systemic T-regulatory responses.
Additionally, of some importance, is the observation that eventually most forms of cancer therapies, regardless of their initial proposed mode(s) of action, are later found to have some direct action on, or through, the immune system, now described with virtually all forms of cancer therapy.
So, if this is true, then why is the immune response driven in the correct direction in some cases to induce CRs, but not in other cases? The answer to this may likely lie in the recent reports of the “bimodality” of action of various and numerous cytokines and chemokines within the human and animal immune systems,Citation30–Citation34 and in the “time-dependent” dynamic nature of the immune response. An important hint at what is likely occurring within our immune systems in cancer is the IL-2 and IL2R interaction in a basic acute immune response. Antigen is presented; IL2R expression increases initially on T-effector cells; these are stimulated by released endogenous IL2; IL2R levels then decrease on T-effector cells and are lost from their surface; T-effector cells then expand rapidly. As part of a feedback response loop some 48-hours later, T-regulatory cells commence expression of IL2R on their surface; these are then in turn stimulated by released IL2; IL2R levels then decrease on T-regulatory cells and are lost from their surface; T-regulatory cells then expand rapidly. After a refractory delay of some 3–5 days, the cycle repeats itself until antigen is removed from the system. If antigen is removed completely during the first cycle, an “acute immune response” is all that is detected clinically; however, if antigen persists – as is inevitably the case with advanced nonresectable cancer – the cycle repeats itself continuously as a “chronic immune response.” If cancer can be removed, then the cycle abates and is damped almost completely; however, if cancer remains, then the inflammatory cycle continuously attenuates itself to produce a situation of tolerance for the aberrant antigen source. This type of repeating inflammatory cycle in advanced cancer patients has been documented and described now in three clinical laboratories internationally, and has been observed to disappear with surgical resection of the cancer. Detection of the cycle is dependent on repeated, serial, near-daily measurements to define cyclical fluctuations in the inflammatory markers. The cycle periodicity appears to be about 6–7 days, with certain defined therapeutic windows of about 12 hours duration for potential therapeutic effectiveness. Citation6–Citation9 There appears to be a separate 12-hour window in each repeating cycle providing an opportunity for inducing activation (responsiveness or stimulation), and another for inducing inhibition (tolerance or regulation). If the inflammatory stimulus, from cell injury or death for example, is generated within the correct critical 12-hour window during the approximate 7-day cycle, then the immune response can be driven in the desired direction for increased responsiveness in order to drive the preexisting already occurring (but weak and ineffective) anticancer immune response more strongly to overcome problematic coexisting homeostatic regulatory/inhibitory/tolerogenic responses. These observations and points may help answer why multiple forms of apparently divergent therapy can damage cancer cells, and in some cases effectively enhance existing clinical immune responses to induce complete cancer regression, but currently, in a random and unpredictable manner.Citation6–Citation9
Summary
Multiple forms of cancer therapy, including pure immunological therapies, propose widely different mechanisms of action for creating CRs and clinical successes. In this review and opinion article some of the critical questions that may better explain this apparent paradox have been explored. Many therapies cause cell damage, cell killing, cancer cell antigen release, danger signals, inflammatory activation, and immune system stimulation ( below).Citation5,Citation10 In this way, micromanipulation of the human immune system in patients with advanced cancer is extremely likely to be actively occurring with most, if not all, forms of past and current cancer therapies in vivo. This effect is evident through direct damage or ablation of immunological cell populations altering the relative responsiveness–tolerance balance, and/or from the release of tumor antigens from cancer cell injury causing enhanced immunostimulation of existing immune responses against the cancer. If these points are clarified, treatments for cancer might be targeted more specifically for greatly improved clinical efficacy in terms of morbidity, CRs, and patient survival.
Figure 1 Multiple cancer therapies leading to a final common pathway of action.
Notes: Many, if not all, cancer therapies lead to cancer cell damage, antigen release, and danger signals, and a final common pathway of immunomodulation, causing in vivo responsiveness or tolerance to advanced cancer inducing cancer regression or growth, respectively.
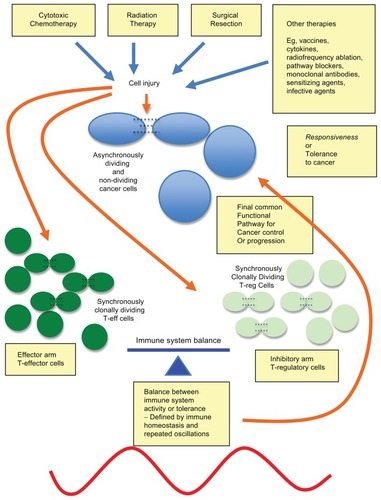
When it is fully appreciated that it is immune system micromanipulation that can lead to random CRs, then it is highly likely that this can be harnessed for predictable control of cancer and chronic inflammation using significant therapeutic strategies based on these approaches.Citation6–Citation13
The present authors’ reasoning for a “final common pathway” through the human inflammatory and immune system offers a quite valid explanation for the observations over time for the random, consistently low number of therapeutic successes from multiple, very seemingly divergent, forms of cancer therapies. Moreover, the time-course of therapy with regard to the patient’s own individual immune response (requiring frequent regular near-daily monitoring to detect) is likely to be a pivotal and critical missing factor in determining the direction that the immune response will ultimately be driven by therapeutic interventions to achieve control of cancer (and potentially other states of chronic inflammation) and to improve the clinical outcome. These points have until now, it appears, been largely “lost in translation.”
Acknowledgments
The authors would like to thank Prof S Markovic, Mayo Clinic, MN; Prof A Dalgleish, St George’s Hospital, London; Prof M Lotze, Pittsburgh Cancer Institute, PA; Prof R Hawkins, Christie Hospital, Manchester, UK; Prof D Morton, John Wayne Cancer Institute, Santa Monica, CA; Prof A Cochran, Pathology, UCLA; and Dr R Neves and Dr J Drabick, Hershey Medical Centre, Hershey, PA, for broad and helpful discussions.
Disclosure
The authors declare no conflicts of interest with regard to the content of this paper.
References
- KiberstisPATravisJCelebrating a glass half-fullScience20063121157
- MurphyKMTopelRHThe value of health and longevityJ Polit Econ2006114871904
- US National Institues of HealthPresident’s Cancer Panel2007–2008 annual report Available from: http://deainfo.nci.nih.gov/advisory/pcp/pcp.htmAccessed April 23, 2012
- MorganGWardRBartonMThe contribution of cytotoxic chemotherapy to 5-year survival in adult malignanciesClin Oncol (R Coll Radiol)20041654956015630849
- DeVitaVTJrLawrenceTSRosenbergSADePinhoRAWeinbergRADeVita, Hellman, and Rosenberg’s Cancer: Principles and Practice of Oncology9th edNew YorkLippincott2011
- CoventryBJAshdownMLQuinnMAMarkovicSNYatomi-ClarkeSLRobinsonAPCRP identifies homeostatic immune oscillations in cancer patients: a potential treatment targeting tool?J Transl Med2009710219948067
- AshdownMLCoventryBJA Matter of TimeAustralasian Science520101820
- CoventryBJImmuno-chemotherapy using repeated vaccine treatment can produce successful clinical responses in advanced metastatic melanomaJ Cancer Ther20101205213
- LeontovichAADroncaRSSumanVJFluctuation of systemic immunity in melanoma and implications for timing of therapyFront Biosci (Elite Ed)2012495897522201928
- LeslieM‘Danger signals’ from dying cells jolt immune system into actionScience Epub February 9, 2012
- LugadeAAMoranJPGerberSARoseRCFrelingerJGLordEMLocal radiation therapy of B16 melanoma tumors increases the generation of tumor antigen-specific effector cells that traffic to the tumorJ Immunol2005174127516752315944250
- SchaueDRatikanJAIwamotoKSMcBrideWHMaximizing tumor immunity with fractionated radiationInt J Radiat Oncol Biol Phys Epub December 28, 2011
- TabachnykMDistelLVBüttnerMRadiochemotherapy induces a favourable tumour infiltrating inflammatory cell profile in head and neck cancerOral Oncol Epub February 20, 2012
- OlivierKRSchildSEMorrisCGBrownPDMarkovicSNA higher radiotherapy dose is associated with more durable palliation and longer survival in patients with metastatic melanomaCancer20071101791179517721993
- MortonRTreatment of Metastatic MelanomaCroatiaIntech Publishers2011
- PetersenRPHanishSIHaneyJCJ Thorac Cardiovasc Surg2007133110411017198792
- EssnerRLeeJHWanekLAItakuraHMortonDLContemporary surgical treatment of advanced-stage melanomaArch Surg20041399961966 discussion 966–96715381613
- BalchCHoughtonASoberASoongSCutaneous Melanoma4th edSt LouisQuality Medical Publishing, Inc2003
- OllilaDWHsuehECSternSLMortonDLMetastasectomy for recurrent stage IV melanomaJ Surg Oncol199971420921310440757
- BarthAWanekLAMortonDLPrognostic factors in 1,521 melanoma patients with distant metastasesJ Am Coll Surg199518131932017670677
- WongJHEuhusDMMortonDLSurgical resection for metastatic melanoma to the lungArch Surg19881239109110953415459
- LoiSde AzambujaEPuglianoLSotiriouCPiccartMJHER2- overexpressing breast cancer: time for the cure with less chemotherapy?Curr Opin Oncol201123654755821918439
- MurphyCGFornierMHER2-positive breast cancer: beyond trastuzumabOncology (Williston Park)201024541041520480738
- AbramsonVArteagaCLNew strategies in HER2-overexpressing breast cancer: many combinations of targeted drugs availableClin Cancer Res201117595295821248299
- GrantSLHammacherADouglasAMAn unexpected biochemical and functional interaction between gp130 and the EGF receptor family in breast cancer cellsOncogene (England)2002213460474
- HalabanRZhangWBacchiocchiAPLX4032, a selective BRAF(V600E) kinase inhibitor, activates the ERK pathway and enhances cell migration and proliferation of BRAF(WT) melanoma cellsPigment Cell Melanoma Res201123219020020149136
- ErnstoffMSBeen there, not done that – melanoma in the age of molecular therapyN Engl J Med2011364262547254821639809
- FlahertyKTPuzanovIKimKBInhibition of mutated, activated BRAF in metastatic melanomaN Engl J Med201036380981920818844
- ChapmanPBHauschildARobertCImproved survival with vemurafenib in melanoma with BRAF V600E mutationN Engl J Med2011364262507251621639808
- RibasAFlahertyKTBRAF targeted therapy changes the treatment paradigm in melanomaNat Rev Clin Oncol20118742643321606968
- SosmanJAKimKBSchuchterLSurvival in BRAF V600-mutant advanced melanoma treated with vemurafenibN Engl J Med2012366870771422356324
- HatzivassiliouGSongKYenIRAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growthNature2010464728743143520130576
- GrivasPDRedmanBGImmunotherapy of kidney cancerCurr Clin Pharmacol20116315116321827390
- KnoechelBLohrJKahnEBluestoneJAAbbasAKSequential development of interleukin 2-dependent effector and regulatory T cells in response to endogenous systemic antigenJ Exp Med2005202101375138616287710
- McNallyAHillGRSparwasserTThomasRSteptoeRJCD4+CD25+ regulatory T cells control CD8+ T-cell effector differentiation by modulating IL-2 homeostasisProc Natl Acad Sci U S A2011108187529753421502514
- SojkaDKHughsonASukiennickiTLFowellDJEarly kinetic window of target T cell susceptibility to CD25+ regulatory T cell activityJ Immunol2005175117274728016301632
- AhmadzadehMRosenbergSAIL-2 administration increases CD4+ CD25(hi) Foxp3+ regulatory T cells in cancer patientsBlood200610762409241416304057
- CoventryBJAshdownMLMarkovicSNImmune therapies for cancer: bimodality – the blind spot to clinical efficacy – lost in translationJ Immunother2011349717
- BitonJBoissierMCBessisNTNFα: activator or inhibitor of regulatory T cells?Joint Bone Spine201279211912322209316
- FilippiCMJuedesAEOldhamJETransforming growth factor-beta suppresses the activation of CD8+ T-cells when naive but promotes their survival and function once antigen experienced: a two-faced impact on autoimmunityDiabetes200857102684269218689691
- O’ConnorWJrKamanakaMBoothCJA protective function for interleukin 17 in T cell-mediated intestinal inflammationNat Immunol200910660360919448631
- ChenXOppenheimJJTNF-alpha: an activator of CD4+FoxP3+ TNFR2+ regulatory T cellsCurr Dir Autoimmun20101111913420173391
- EvangelidouMTsevelekiVVamvakasSSProbertLTNFRI is a positive T-cell costimulatory molecule important for the timing of cytokine responsesImmunol Cell Biol201088558659520212506
- ChenXOppenheimJJContrasting effects of TNF and anti-TNF on the activation of effector T cells and regulatory T cells in autoimmunityFEBS Lett2011585233611361821513711
- ComerfordIBuntingMFenixKAn immune paradox: how can the same chemokine axis regulate both immune tolerance and activation?: CCR6/CCL20: a chemokine axis balancing immunological tolerance and inflammation in autoimmune diseaseBioessays201032121067107620954179
- MummJBEmmerichJZhangXIL-10 elicits IFNγ-dependent tumor immune surveillanceCancer Cell201120678179622172723
- A delicate balance: tweaking IL-2 immunotherapyNat Med201218220820922310686
- PrietoPAYangJCSherryRMCTLA-4 blockade with ipilimumab: long-term follow-up of 177 patients with metastatic melanomaClin Cancer Res20121872039204722271879
- JainNNguyenHChambersCKangJDual function of CTLA-4 in regulatory T cells and conventional T cells to prevent multiorgan autoimmunityProc Natl Acad Sci U S A20111074152420080649
- HodiFSO’DaySJMcDermottDFImproved survival with ipilimumab in patients with metastatic melanomaN Engl J Med201036371172320525992
- QureshiOSZhengYNakamuraKTrans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4Science201129:332602960060321474713
- CoventryBHeinzelSCD1a in human cancers: a new role for an old moleculeTrends Immunol200425524224815099564
- XuHCaoXDendritic cell vaccines in cancer immunotherapy: from biology to translational medicineFront Med20115432333222198743
- PandolfiFCianciRPagliariDThe immune response to tumors as a tool toward immunotherapyClin Dev Immunol2011201189470422190975
- TuyaertsSDendritic cell therapy for oncology roundtable conferenceJ Immune Based Ther Vaccines201191121226916
- NakaiNHartmannGKishimotoSKatohNDendritic cell vaccination in human melanoma: relationships between clinical effects and vaccine parametersPigment Cell Melanoma Res201023560761920579307
- TumaRSLarge trials, small gains: is change on the way?J Natl Cancer Inst20101021612161217122320693460
- GlundeKBhujwallaZMMetabolic tumor imaging using magnetic resonance spectroscopySemin Oncol2011381264121362514
- GlundeKJiangLMoestueSAGribbestadISMRS and MRSI guidance in molecular medicine: targeting and monitoring of choline and glucose metabolism in cancerNMR Biomed201124667369021793073