
Abstract
Metabolic alteration, one of the hallmarks of cancer cells, is important for cancer initiation and development. To support their rapid growth, cancer cells alter their metabolism so as to obtain the necessary energy and building blocks for biosynthetic pathways, as well as to adjust their redox balance. Once thought to be merely byproducts of metabolic pathways, intermediate metabolites are now known to mediate epigenetic modifications and protein post-transcriptional modifications (PTM), as well as connect cellular metabolism with signal transduction. Consequently, they can affect a myriad of processes, including proliferation, apoptosis, and immunity. In this review, we summarize multiple representative metabolites involved in glycolysis, the pentose phosphate pathway (PPP), the tricarboxylic acid (TCA) cycle, lipid synthesis, ketogenesis, methionine metabolism, glutamine metabolism, and tryptophan metabolism, focusing on their roles in chromatin and protein modifications and as signal-transducing messengers.
Introduction
Cell metabolism comprises an intricate network of chemical reactions that sustain normal growth and reproduction. Metabolism comprises catabolism and anabolism, the former supplying energy and the latter producing the necessary cellular components for cell proliferation. Cancers are characterized by uncontrolled cell proliferation and heterogeneous microenvironment. On the one hand, cancer cells adjust their metabolic preference to balance their energy needs with the need to generate biosynthetic precursors for growth;Citation1 on the other hand, they develop nutrient-scavenging strategies to survive under nutrient-starvation and oxygen-limiting conditions.Citation2 Cancer cells undergo extensive metabolic alterations, including in glycolysis, mitochondrial biogenesis, lipid metabolism, and the pentose phosphate pathway (PPP),Citation3 by either reprogramming the activities of existing metabolic pathways or rewiring new connections.Citation4
Metabolic reprogramming in cancer cells results in the accumulation or depletion of intermediate metabolites through a variety of mechanisms.Citation5 The first one is the alteration of metabolic enzyme activity. For example, during glycolysis, the preferred way for cancer cells to obtain energy and biosynthetic building blocks, the activation of glycolysis-related enzymes leads to the accumulation of a series of glycolytic intermediates.Citation6 In contrast, the loss of the activities of succinate dehydrogenase (SDH) and fumarate hydratase (FH) contributes to the accumulation of succinate and fumarate, respectively.Citation7 Secondly, mutations arising in cancer cells can result in neomorphic enzyme activity. For instance, wild-type isocitrate dehydrogenase (IDH) converts isocitrate to alpha-ketoglutarate (α-KG), while IDH with specific single-site mutations further catalyzes the conversion of α-KG to 2-hydroxyglutarate (2-HG).Citation8 Thirdly, cancer cells generate several active by-products of metabolic pathways, such as reactive oxygen species (ROS), NAD+/NADH, and NADP+/NADPH.
Since the discovery of the oncogenic roles of some mitochondrial metabolites, such as 2-HG, succinate, and fumarate, research has increasingly focused on investigating the roles of these “oncometabolites” in cancer.Citation9,Citation10 Oncometabolites affect processes such as epigenetic modifications, post-transcriptional modifications (PTMs), and signaling transduction.Citation10 Metabolic remodeling can promote DNA hypermethylation and histone hyperacetylation, thereby silencing tumor suppressor genes and promoting tumorigenesis.Citation11 As for PTMs, a wide spectrum of metabolites can conjugate to proteins and regulate their functions. Various types of PTMs have been reported, among which acetylation and succinylation have attracted extensive research interest.Citation12 To illustrate metabolite sensing and signaling, Wang and colleagues proposed a ternary model consisting of a sensor, a transducer, and an effector.Citation13 In their model, metabolites were first recognized by sensors. Transducers subsequently transmitted the signal information to effectors, which finally stimulated the corresponding biological reactions.Citation13 They grouped a variety of metabolite sensing events into three modes: metabolite sensor-mediated signaling (MeSr), metabolite-sensing module (MeS), and sensing by conjugating (SC).Citation13 In the first category, a sensor physically interacts with the metabolite and transduces the signals to downstream. In the metabolite-sensing module, molecules like protein complexes are disrupted by the metabolites without direct binding through a structurally conserved site, causing downstream changes. The last mode is the conjugation of metabolites to proteins or nucleotides, causing functional alterations.Citation13
Besides oncometabolites, numerous intermediate metabolites can directly bind to proteins or nucleotides, leading to their dysfunction. In addition, these intermediate metabolites can act as ligands for transmembrane receptors, activating downstream signaling cascades. In this review, we will introduce the roles of multiple intermediates classified by metabolic pathways in cancer. For each intermediate metabolite, we will briefly introduce its source, and then discuss in-depth its effects on epigenetic modifications, PTM, and signaling transduction ().
Table 1 Roles of Metabolites in Chromatin and Protein Modifications, Signal Transduction and Their Effects on Cancer
Metabolites in Glycolysis
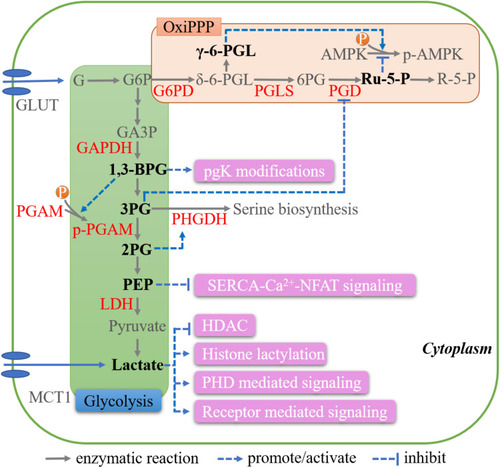
Glycolysis consists of energy-requiring and energy-releasing phases.Citation14 In the first step of the energy-requiring phase, hexokinase catalyzes the phosphorylation of glucose, generating glucose-6-phosphate (G6P). G6P is then transformed to glyceraldehyde-3-phosphate (GA3P) through several steps. GA3P is oxidized to 1,3-bisphosphoglycerate (1,3-BPG) by glyceraldehyde 3-phosphate dehydrogenase (GAPDH), which is the first step in the energy-releasing phase. 1,3-BPG loses a phosphate and becomes 3-phosphoglycerate (3-PG), which is further converted to 2-phosphoglycerate (2-PG) by phosphoglycerate mutase (PGAM). After losing one molecule of H2O, 2-PG is converted to phosphoenolpyruvate (PEP). Dephosphorylation of PEP yields pyruvate. Under oxygen-rich conditions, pyruvate is transferred to the mitochondria and participates in the tricarboxylic acid (TCA) cycle; under hypoxic conditions; however, pyruvate is converted to lactate by lactate dehydrogenase (LDH)Citation14 ().
Figure 1 Key metabolites in glycolysis and oxidative PPP. Schematic representations of the biologic effects of those metabolites, including 1,3-BPG, 3PG, 2PG, PEP, lactate, γ-6-PGL and Ru-5-P, are in pink.
1,3-BPG
1,3-BPG brings about PTMs to a variety of glycolytic proteins. During the modification process, active 1,3-BPG binds to lysine residues in these proteins, generating 3-phosphoglyceryl-lysine (pgK) in an enzyme-independent manner. Under high-glucose conditions, the generation of pgK inhibits glycolysis and redirects glycolytic intermediates to alternative biosynthetic pathways, which represents a crucial feedback regulatory mechanism (SC mode).Citation15 Additionally, 1,3-BPG can activate PGAM1 by directly phosphorylating its histidine residues (), thereby maintaining glycolytic flux and supporting cell growth in HCT116 or MDA-MB-231 cancer cells.Citation16
3-PG and 2-PG
3-PG competitively occupies the active site of phosphogluconate dehydrogenase (PGD), the rate-limiting enzyme in the PPP, resulting in impaired PGD function and the suppression of the PPP fluxCitation17 (). Increased expression of PGAM1 in tumor cells leads to enhanced 3-PG consumption, PPP activation, and increased 2-PG levels. 2-PG can further downregulate the level of 3-PG by enhancing the phosphoglycerate dehydrogenase (PHGDH)-mediated production of 3-phosphohydroxypyruvate (pPYR) (), which is the first committed step in serine synthesis. Through these mechanisms, tumor cells precisely adjust 3-PG and 2-PG levels, thereby regulating glycolysis and anabolic biosynthesis.Citation17
PEP
In intratumoral T cells, PEP suppresses the activity of sarco/endoplasmic reticulum Ca(2+)-ATPase (SERCA), a calcium transporter that mediates Ca2+ uptake into the endoplasmic reticulum, resulting in Ca2+ accumulation in the cytosol and further nuclear factor of activated T cells (NFAT) signaling activation, which is vital for T cells to exert their antitumor effects.Citation18,Citation19 This process of metabolite sensing and signaling can be classified as the MeS mode.
Lactate
Due to higher glucose-to-lactate flux, lactate overproduction is commonly detected in a subset of cancer cells, especially under hypoxic conditions. In oxygenated tumor cells, monocarboxylate transporter 1 (MCT1) can also mediate lactate import from hypoxic cancer cellsCitation20,Citation21 ().
Although widely known as an energy source, lactate displays active nonmetabolic characteristics. Lactate can inhibit α-KG-dependent prolyl hydroxylase domain proteins (PHDs), which are involved in the hydroxylation of hypoxia-inducible factor 1-alpha (HIF-1α) and IκB kinase β (IKKβ). Inhibition of HIF-1α leads to its stabilization, leading to the activation of HIF-1-mediated vascular endothelial growth factor (VEGF) signaling.Citation22 Meanwhile, the inhibition of IKKβ hydroxylation results in IKBα degradation, which activates nuclear factor kappa B (NF-κB) signaling.Citation23 The effects of lactate on VEGF and NF-κB signaling can be classified as the MeS mode. Lactate can also interrupt the association between PHDs and N-Myc downstream-regulated protein (NDRG3) by directly binding to the latter in a manner that is independent of HIFs, which is an MeSr mode. This prevents the proteasomal degradation of NDRG3 and further activates Raf/ERK signaling, contributing to angiogenic and proliferative effects in cancer cells.Citation24
In terms of driving immune evasion, the lactate-induced expression of arginase 1 (Arg1) promotes the functional polarization of tumor-associated macrophages (TAMs).Citation25,Citation26 In breast cancer, lactate was reported to induce TAM polarization through the ERK/STAT3 pathway.Citation27 There are several examples of MeSr mode about lactate driving immune evasion. G protein-coupled receptor 81 (GPR81) is a lactate receptor that is highly expressed in multiple cancer cell lines.Citation28 Lactate-GPR81 signaling stimulates the expression of programmed cell death ligand 1 (PD-L1) through the transcriptional coactivator TAZ, thereby suppressing interferon-gamma production in lung cancer.Citation29,Citation30 Lactate also suppresses innate immune responses in cancer. By binding to GPR81, lactate inactivates yes-associated protein (YAP) and further disrupts the interaction of YAP and NF-κB in macrophages, resulting in the reduced production of macrophage pro-inflammatory cytokine.Citation31 Furthermore, Zhang et al reported that lactate prevented the aggregation of mitochondrial antiviral-signaling protein (MAVS) by directly binding to its transmembrane domain. This suppressed the production of downstream type I interferons triggered by retinoic acid-inducible gene I-like receptor (RLR)-MAVS signaling, impairing cancer immunosuppression.Citation32
Lactate also regulates gene expression by inhibiting histone deacetylases (HDACs),Citation33 and can provide the lactyl group for lysine residues in histone tails, known as histone lactylation. Histone lactylation is active in TAMs, implying that this process has a role in immune surveillance.Citation34
Metabolites in the PPP
The PPP consists of oxidative and nonoxidative phases, and leads to the production of metabolites and NADPH, which are pivotal for nucleotide biosynthesis, lipogenesis, and the maintenance of redox homeostasis. In the first step, G6P is oxidized to δ-6-phosphogluconolactone (δ-6-PGL) by glucose-6-phosphate dehydrogenase (G6PD). Then, one carbon of hydrolytically unstable δ-6-PGL is cleaved by 6-phosphogluconolactonase (PGLS), yielding 6-phosphogluconate (6-PG). 6-PG is further converted to ribulose-5-phosphate (Ru-5-P) by PGD. Ru-5-P is isomerized into ribose-5-phosphate (R-5-P), which serves as the main building block for ribonucleotide synthesis.Citation35 Notably, there is another form of 6-PGL—γ-6PGL—that is generated by the intramolecular rearrangement of δ-6PGL. It is relatively stable, represents a “dead-end” byproduct, and is not subsequently involved in the PPP ().
Ru-5-P and 6-PGL
AMP-activated protein kinase (AMPK), a central metabolic sensor, is activated by upstream kinases and inactivated by phosphatase-mediated dephosphorylation. Liver kinase B1 homolog (LKB1) can form a complex with STE20-related adaptor protein (STRAD) and mouse protein 25 (MO25), acting as a major upstream activator of AMPK.Citation36 Protein phosphatase 2A (PP2A) is a serine/threonine protein phosphatase that dephosphorylates AMPK at Thr172, thereby inactivating it.Citation37 Ru-5-P can disrupt the LKB1-MO25-STRAD complex, resulting in the inactivation of AMPK.Citation38 In contrast, γ-6PGL binds to Src and inhibits PP2A activity by dephosphorylation, leading to AMPK activationCitation39 (). The action of Ru-5-P and γ-6PGL belongs to the MeS and MeSr mode, respectively. Cancer cells exhibit an active oxidative PPP, accompanied by decreased γ-6PGL and increased Ru-5-P levels. These alterations collectively inactivate AMPK, activate acetyl-CoA carboxylase 1, and, finally, enhance lipogenesis and tumor growth.Citation38,Citation39
Metabolites in the TCA Cycle
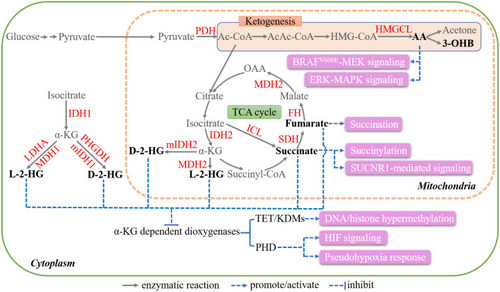
The TCA cycle, also called the Krebs cycle or the citric acid cycle, comprises a series of enzyme-catalyzed reactions and is the major energy production pathway in cells.Citation40 The third step is catalyzed by IDH, in which isocitrate undergoes oxidation to form α-KG, releasing NADH. α-KG is further converted to succinate, which is enzymatically catalyzed to fumarate by SDH. Fumarate is further oxidized to malate by FH. Finally, malate is oxidized to oxaloacetate (OAA) by malate dehydrogenase (MDH). Notably, MDH catalyzes the interconversion of malate and OAA. There are two isoforms of MDH, namely MDH1 and MDH2, which are localized to the cytoplasm and mitochondria, respectively. Similarly, IDH has three isoforms—cytosolic IDH1 as well as mitochondrial IDH2 and IDH3—with IDH3 primarily functioning in normal enzymatic processesCitation40 ().
Figure 2 Key metabolites in TCA cycle and ketogenesis. The blue arrows and pink boxes highlight the extrametabolic functions of 2-HG, succinate, fumarate, AA, and 3-OHB.
2-HG
2-HG consists of two enantiomers, namely D-2-HG (also known as R-2-HG) and L-2-HG (also known as S-2-HG) (). 2-HG has attracted extensive research interest since the discovery of the neomorphic enzymatic activity of mutant IDH (mIDH) in 2009.Citation8,Citation10,Citation41 Specific missense mutations in both IDH1 and IDH2 result in a neomorphic enzymatic activity that catalyzes α-KG to D-2-HG, but not L-2-HG. The most common mutations are Arg132 in IDH1 and Arg172 plus Arg140 in IDH2, occurring in ~80% of low-grade gliomas and ~20% of cases of acute myeloid leukemia (AML),Citation42–Citation44 as well as in a spectrum of other malignancies, including cartilaginous tumors, intrahepatic cholangiocarcinoma, and angioimmunoblastic T cell lymphoma.Citation45,Citation46 In addition to being produced by mIDH in the TCA cycle, 2-HG can also be generated through several promiscuous enzymatic reactions. PHGDH, which normally catalyzes the first step of serine biosynthesis, has been reported to be a source of D-2-HG in human breast cancer cell lines at quite low efficiency.Citation47 In contrast, and surprisingly, L-2-HG is produced through the activities of MDH and lactate dehydrogenase A (LDHA). MDH catalyzes the production of L-2-HG in mammals, although this reaction is 10Citation7,Citation8 times less efficient when compared with OAA production.Citation48 LDHA has been identified as the major L-2-HG-producing enzyme under hypoxic conditionsCitation49 (). D-2-hydroxyglutarate dehydrogenase (D2HGDH)Citation50,Citation51 and L-2-hydroxyglutarate dehydrogenase (L2HGDH)Citation52 catalyze the conversion of D-2-HG and L-2-HG to α-KG, respectively. Mutations in these two enzymes result in 2-HG accumulation and lead to 2-hydroxyglutaric acidurias (2HGAs).Citation50,Citation52
2-HG functions as an antagonist of α-KG as they have highly similar structures.Citation45 Crystallographic structural studies revealed that 2-HG competitively occupies the active binding sites of multiple α-KG-dependent enzymes, including the JmjC domain-containing histone demethylases (KDMs) and the ten-eleven translocation (TET) family of 5-methylcytosine (5mC) hydroxylases, and hence inhibits their activities.Citation53–Citation55 These enzymes remove methyl moieties through sequential reactions, the inhibition of which by 2-HG increases global DNA methylation and epigenetically silences multiple proteins with known and postulated roles in tumor suppressionCitation53 ().
Histone demethylases are classified into two subfamilies, ie, the lysine demethylase 1 (KDM1) subfamily and the JmjC domain-containing KDMs (consisting of over 30 enzymes in humans, including KDM2, KDM3, KDM4, KDM5, KDM6, and others).Citation56 Enzymes in the latter group are the major targets of D-2-HG. The related inhibition potencies vary, with KDM4A/JMJD2A being the most sensitive to D-2-HG, followed by KDM4C/JMJD2C, KDM2A/FBXL11, AlkB homolog 2 (ALKBH2), factor inhibiting HIF (FIH), PHD, and BBOX-1.Citation54 Studies showed that the addition of D-2-HG or the stable overexpression of mIDH1 augments histone demethylation in glioma, including H3K4, H3K9, H3K27, and H3K79, an effect that can be counteracted by α-KG treatment.Citation53,Citation57 D-2-HG activates the mechanistic (previously mammalian) target of rapamycin (mTOR) signaling pathway in brain cancer by inhibiting KDM4A,Citation58 which is an MeSr mode.
TET sequentially converts 5mC first to 5-hydroxymethylcytosine (5hmC), then to 5-formylcytosine, and finally to 5-carboxylcytosine.Citation59 Biochemical assays showed that D-2-HG exerts a direct inhibitory effect on recombinant TET2 (), which can be rescued by α-KG. mIDH expression leads to the consistent reduction of TET2-dependent 5hmC levels.Citation53 Moreover, both mIDH expression and TET2 silencing impair hematopoietic differentiation, implying that they have similar proleukemogenic effects.Citation60
2-HG can also modulate the activity of several α-KG-dependent dioxygenases independently of epigenetic alterations. PHD, which is involved in the hydroxylation of HIF1α, was the first dioxygenase reported to be inhibited by 2-HG (MeSr mode).Citation61,Citation62 The inhibition of hydroxylation leads to HIF-1α stabilization and the subsequent activation of genes containing HIF response elements (HREs), such as glycolytic enzymes, further contributing to tumor development.Citation63 Additionally, D-2-HG has been reported to directly inhibit ALKBH2,Citation54 a protein that repairs DNA damage caused by alkylating agents. Cells with mIDH accumulate double-strand breaks (DSBs) in their DNA, leading to genetic instability.Citation64
Succinate
Succinate accumulation results primarily from loss-of-function mutations in SDH, which are found in a variety of cancer types, such as paraganglioma/pheochromocytoma (PGL/PCC), renal carcinoma, ovarian cancer, neuroblastoma, and gastrointestinal stromal tumor.Citation65,Citation66 Impairment of SDH activity can also lead to succinate accumulation. For example, tumor necrosis factor receptor-associated protein 1 (TRAP1) downregulates SDH activity by inhibiting respiratory complex II.Citation67 Tumor-associated inflammatory responses can also suppress SDH activity, while isocitrate lyase (ICL) also likely contributes, as it directly converts isocitrate to succinateCitation66 ().
Like 2-HG, succinate can competitively inhibit α-KG-dependent KDMs and TETs,Citation68 resulting in epigenetic alterations. Succinate promotes proliferation, epithelial-to-mesenchymal transition (EMT), migration, and invasion by regulating the activities of a plethora of downstream genes.Citation69–Citation72 For example, increased succinate levels due to SDH mutations lead to DNA and histone hypermethylation in PGL/PCC, thereby suppressing EMT and neuroendocrine differentiation.Citation69,Citation73 Succinate also inhibits PHD and impairs HIF-1α signaling (MeSr mode)Citation74,Citation75 ().
Besides, succinate provides a succinyl moiety for lysine succinylation, a PTM that occurs in both chromatin and metabolic enzymes such as GLUT1, LDHA, and GAPDH,Citation76 regulating their activities in cancer (SC mode)Citation10,Citation77,Citation78 (). For example, Li et al proved that succinylation of LDHA decreased its lysosomal degradation, promoting cell proliferation, invasion, and migration in gastric cancer.Citation79
In addition, succinate can activate signaling pathway by the MeSr mode. Succinate binds to GPR91 (also known as succinate receptor 1 [SUCNR1]), which leads to increased VEGF expression and further triggers downstream signaling cascades, including those associated with extracellular regulated kinase (ERK) 1/2 and signal transducer and activator of transcription 3 (STAT3).Citation80 Succinate secreted by cancer cells binds to GPR91, further activating PI3K/HIF-1α signaling and triggering TAM polarization (). This promotes cancer cell migration, invasion, and metastasis.Citation81
Fumarate
Abnormal fumarate accumulation is attributed to inactivating mutations in FH, which have been reported in skin leiomyomata, uterine fibroids, and papillary renal cell cancer.Citation82 Fumarate appears to be multifaceted.Citation10 Like 2-HG and succinate, fumarate can allosterically inhibit α-KG-dependent enzymes, including KDMs, TETs, and PHDs (MeSr mode),Citation68,Citation73 regulating the epigenetic landscape and producing pseudohypoxia. A distinct PTM related to fumarate is succination, during which the thiol group of a cysteine residue is converted to S-(2-succino)-cysteine (2SC)Citation83 (). Fumarate accumulation affects the normal succination of glycolytic enzymes, adiponectin, cytoskeletal proteins, and endoplasmic reticulum chaperones, impairing their functions.Citation84,Citation85 For instance, when succinated, Kelch-like ECH-associated protein-1 (KEAP1) is dissociated from nuclear factor erythroid 2-related factor 2 (NRF2). The latter is then stabilized, translocated to the nucleus, and modulated the transcription of several genes involved in antioxidant signaling and cytoprotection.Citation86,Citation87 In addition, the direct binding of fumarate to glutathioneCitation88 or glutathione succinationCitation89 leads to persistent oxidative stress and cellular senescence.
Metabolites in Lipid Synthesis
Acetyl-CoA
Acetyl-CoA (Ac-CoA) is the main intermediate for lipid synthesis and is located in several cellular compartments, including the cytosol, mitochondria, and the nucleus. Generally, pyruvate is transferred to the mitochondria and decarboxylated to Ac-CoA by pyruvate dehydrogenase (PDH).Citation90 Interestingly, Ac-CoA synthetase (ACSS) is another major enzyme that catalyzes the ATP-dependent incorporation of acetate into Ac-CoA. In mammalian cells, there are two ACSSs, namely, mitochondrial ACSS1 and cytosolic ACSS2. The expression of ACCS1 is markedly upregulated in multiple tumors.Citation91 Under hypoxia, mitochondrial citrate is preferentially shuttled to the cytoplasm and converted to Ac-CoA and OAA by ATP citrate lyase (ACL).Citation92 Acetate uptake, which is catalyzed by ACSS2, is also responsible for increased Ac-CoA levels.Citation93 Reductive glutamine metabolism by IDH1 is another source of Ac-CoA under hypoxic conditionsCitation94 ().
Figure 3 Key metabolites in lipid synthesis. Schematic diagram shows the Ac-CoA pool in different cell compartments. The functions of Ac-CoA and acetate outside metabolism are marked in pink.
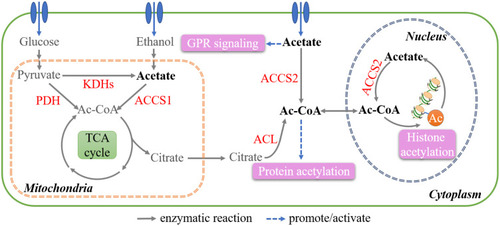
Ac-CoA is the starting material for fatty acid synthesis, while also being indispensable for the acetylation of histones and proteins (SC mode). There is a strong correlation between Ac-CoA levels and global histone acetylation.Citation95,Citation96 By modulating epigenetic alterations, Ac-CoA regulates the expression of numerous genes and ultimately promotes tumorigenesis.Citation97 For example, in pancreatic ductal adenocarcinoma harboring KRAS mutations, ACL-mediated H3K27 acetylation (H3K27ac) is increased, thereby promoting tumor development.Citation98 H3K27ac due to upregulated Ac-CoA level also promotes the progression and chemoresistance of nasopharyngeal carcinoma.Citation99 In human hepatocellular carcinoma, hypoxic cells show increased histone H3 acetylation due to an increase in Ac-CoA levels catalyzed by ACCS, which promotes lipid synthesis and tumor growth.Citation100 Numerous proteins are also modulated through post-translational acetylation. For example, when acetylated at K540, 546, and 554, ACL tends to be stabilized, leading to increased Ac-CoA production through a feedforward mechanismCitation101 (). After K413 acetylation, mutant IDH2 (mIDH2) R140Q presents higher enzyme activity, producing sufficient 2-HG for the transformation in AML.Citation102
Acetate
Exogenous acetate is mainly derived from saccharolytic fermentation in the colon, but can also be obtained from other sources, such as ethanol oxidation. Endogenously, acetate is generated via deacetylation and hydrolysis reactions that release acetyl groups. Recent studies have shown that acetate is produced de novo from pyruvate, via either ROS-dependent oxidative decarboxylation or incomplete oxidation by ketoacid dehydrogenases (KDHs) in a thiamine- and glutathione-dependent mannerCitation103 ().
As previously mentioned, acetate maintains the Ac-CoA pool in cancer cells, thereby influencing multiple Ac-CoA-associated biological processes.Citation104 Acetate is also an agonist of free fatty acid receptors (FFARs), which belong to the GPR family. Acetate activates FFAR2 in 3T3 fibroblasts, potentiating their malignant transformation.Citation105 In breast cancer, acetate-GPR signaling, which belongs to the MeSr mode, activates p38 mitogen-activated protein kinase (MAPK) and, subsequently, heat shock protein 27 (HSP27), thereby preventing stress-induced damageCitation106 ().
Long Chain Fatty Acids (LCFA)
Besides the role of energy source, LCFA also functions as signaling molecules. By displacing retinoic acid, saturated LCFA (SLCFA) binds to and inhibits fatty acid-binding protein 5 (FABP5) (MeSr mode). This suppresses the nuclear localization of peroxisome proliferator-activated receptor β/δ (PPARβ/δ), inhibiting the growth of carcinoma cells in vitro and in vivo.Citation107,Citation108 Interestingly, unsaturated LCFA (ULCFA) displays opposing effects.Citation107
Metabolites in the Ketogenesis Pathway
In a fasted state, the body switches to breaking down fatty acids to ketone bodies (ketogenesis) to satisfy the energy requirements of key organs, including the brain, muscles, and other tissues. Ketogenesis occurs primarily in the mitochondria and begins with the condensation of Ac-CoA to acetoacetyl-CoA (AcAc-CoA). Subsequently, 3-hydroxy-3-methylglutaryl–CoA synthase (HMGCS) catalyzes the condensation of Ac-CoA and AcAc-CoA to form 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA), which is further cleaved to acetoacetate (AA) by 3-hydroxy-3-methylglutaryl-CoA lyase (HMGCL). AA is then either dehydrogenized to 3-hydroxybutyrate (3-OHB) or decarboxylated to acetone. Finally, AA, 3-OHB, and AcAc-CoA acetone are transferred to the circulation and taken up by cells as alternative energy sourcesCitation109 ().
AA and 3-OHB
AA has been reported to activate ERK1/2 and p38 MAPK signaling in primary cultured rat hepatocytes in a ROS- and oxidative stress-dependent mannerCitation110 (). Kang et al conducted a systematic screen for metabolic synthetic lethal partners of BRAFV600E and found that HMGCL and HMGCS1 could specifically promote the growth of BRAFV600E-positive melanoma.Citation111 The authors further demonstrated that AA bound to BRAFV600E, which enhanced the binding of BRAFV600E and MEK1, thereby promoting the activation of MEK-ERK signaling (MeSr mode).Citation111,Citation112 Meanwhile, a different study reported that a high-fat ketogenic diet could increase serum AA concentrations, resulting in the enhanced growth of tumors derived from BRAFV600E -positive melanoma cells in xenografted mice.Citation113 Conversely, both reducing circulating AA levels with hypolipidemic agents and treating with an inhibitory AA homolog could effectively attenuate the growth of BRAFV600E-positive tumors.Citation113 Collectively, these findings suggest that AA mediates the crosstalk between the ketogenesis metabolic pathway and the MAPK signaling pathway ().
Of note, 3-OHB is known to have functions beyond metabolism, including inhibiting HDACs and transducing signals through GPRs, effects that are closely correlated with diabetes and lifespan.Citation114–Citation117 Similarly, 3-OHB could inhibit Class I HDACs in cancer cells, which increases histone acetylation.Citation118 Moreover, the generation of lysine β-hydroxybutyrylation (Kbhb), a novel type of histone posttranslational modification, is specifically attributed to 3-OHB. This new epigenetic regulatory is enriched in active gene promoters and closely linked with gene expression.Citation119 For example, p53 is a well-known tumor suppressor gene. Its activity was significantly attenuated after Kbhb modification, leading to reduced cell growth arrest and apoptosis in cancer cells.Citation120
As an intracellular signal mediator, 3-OHB also works as the only endogenous ligand of G-protein coupled receptors 109A (GPR109A), which is a potent tumor suppressor. In colonic epithelial cells, 3-OHB activates GPR109A, enhancing colonic cancer cells apoptosis and depressing survival (MeSr mode).Citation121
Metabolites in Methionine Metabolism
S-Adenosyl-Methionine
S-adenosyl-methionine (SAM) is produced from methionine through the activity of methionine adenosyltransferase 2A (MAT2A), which represents the first step in the methionine cycle. In the following step, which is catalyzed by methyltransferases (MTs), SAM donates the methyl group and is converted to S-adenosyl-homocysteine (SAH)Citation122 ().
Figure 4 Key metabolites in methionine and glutamine metabolism. The metabolic reactions closely related to SAM and GSH production are shown. SAM is a universal methyl donor and primarily regulates methylation. GSH mainly modify glutathionylation through binding to the cysteine residues of proteins.
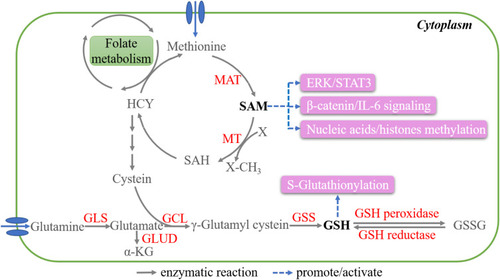
As a universal methyl donor, SAM can greatly influence the methylation status of nucleic acidsCitation123 and histones,Citation124 thereby modulating diverse and critical cellular processes in cancer (SC mode). For example, SAM inhibits the expression of urokinase-type plasminogen activator (uPA) and matrix metalloproteinase-2 (MMP-2) through the hypermethylation of their promoters, which suppresses invasiveness and tumorigenesis in prostateCitation125 and breast cancer.Citation126 Moreover, SAM can reverse the hypomethylation of the promoters of the oncogenes c-Myc and H-Ras, resulting in the inhibition of cell growth in gastric and colon cancer.Citation127
SAM is also reported to reduce inflammation by repressing β-catenin and interleukin-6 (IL-6) signaling in liver and colon cancer,Citation128,Citation129 and also exerts proapoptotic effects through the ERK1/2 and STAT3 pathways in osteosarcoma cellsCitation130 (). However, further studies are needed to elucidate the mechanism underlying its role in signaling transduction.
Glutamine Metabolism
Glutathione
Glutamine is another substrate that is vital for energy production and macromolecule biosynthesis in cancer cells. Glutamine is transported into cells and converted to glutamate by mitochondrial glutaminases (GLSs). Glutamate has two major metabolic fates. It can either be converted to α‑KG by glutamate dehydrogenase (GLUD) or aminotransferases and used for energy production in the TCA cycle or is first converted to γ-glutamylcysteine by glutamate-cysteine ligase (GCL) and then to glutathione (GSH) by GSH synthetase (GSS).Citation131 GSH exists in both thiol-reduced and disulfide-oxidized forms, namely, GSH and GSSG, respectively. GSH can be oxidized to GSSG by GSH peroxidase, while GSSG can be reverse-catalyzed to GSH by GSH reductaseCitation132 ().
GSH is a well-known antioxidant. Abundant GSH in tumor cells protects themselves through the detoxification of carcinogens and the scavenging of free radicals. In terms of PTM, GSH binds to the cysteine residues of proteins, a process called S-glutathionylation, thereby protecting them from ROS attack. A growing number of GSH protein targets have been identified, including P53, HSP27, thioredoxin, caspase-3, and NF-κB, involving a vast number of cellular processes (SC mode).Citation133–Citation135 The S-glutathionylation of histone H3, a GSH target, can lead to altered chromatin structure and nucleosome instability.Citation136
Tryptophan Metabolism
Tryptophan (Trp) is an essential amino acid. A small fraction of Trp enter either serotonin or indole pathway, which are mainly occurred in nervous or innate immune system, respectively. Over 95% of free Trp is degraded by the kynurenine (Kyn) pathway (KP), which is closely related to cancer progression. In the KP, Trp is firstly converted to N-formylkynurenine by indoleamine-2,3-dioxygenase 1 (IDO1), IDO2, and tryptophan-2,3-dioxygenase (TDO), which is the rate-limiting step. N-formylkynurenine is then catalyzed by kynurenine formamidase (AFMID) to produce Kyn. Kyn is further converted to 3-hydroxykynurenine (3-HK) by kynurenine 3-monooxygenase (KMO), to anthranilic acid (AA) by kynureninase (KYNU), and to kynurenic acid (KA) by kynurenine aminotransferases (KATI–KATIII). After further series of enzymic reactions, multiple biologically active acids are produced, such as 3-hydroxyanthranilic acid (3-HAA), quinolinic acid (QA), picolinic acid (PA), etc. In the end of the KP, NAD+ is generated, which is an important redox cofactorCitation137,Citation138 ().
Figure 5 Key metabolites in the tryptophan (Trp) metabolism. The enzymatic reactions in kynurenine (Kyn) pathway (KP) are shown. Kyn and KA work as signal messengers, which are highlighted.
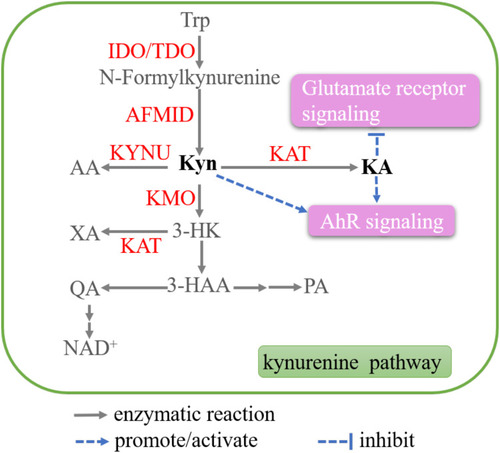
Kyn and KA
Both Kyn and KA are potent agonists for the human aryl hydrocarbon receptor (AhR),Citation139 which has extensive roles in carcinogenesis.Citation140 Thus, Kyn and KA have potential pro-carcinogenic effects in cancer. Kyn is significantly elevated in colon cancer cells and promotes the proliferation through activating the AhR.Citation141 AhR blockade induces by Kyn could also interrupt the interplay between Tregs and tumor-associated macrophages, which is associated with the resistance to immune checkpoint inhibitors.Citation142 DiNatale et al reported that KA activated AhR and subsequently induced IL-6 production in primary human hepatocytesCitation143 ().
KA also works as the ligand of other receptors, including glutamate receptors, α-7 nicotinic acetylcholine receptor (α-7 nAChR), and G-protein coupled receptor 35 (GPR35).Citation144 As antagonist for endogenous glutamate receptors, KA reverses the promotion effect of glutamate on glioma T98G cell proliferation, and enhances the antiproliferative effect of glutamate receptor antagonists MK801 and GYKI 52466Citation145 (). However, the anti-cancer potential of KA through binding to α-7 nAChR and GPR35 warrants further study.
Conclusions and Future Perspectives
Metabolites are multifaceted in cancer cells, exerting metabolic as well as extra-metabolic functions. The comprehensive deciphering of these functions holds immense potential for developing new classes of therapeutics. Multiple intermediates exert extra-metabolic effects on processes such as epigenetic modifications, PTMs, and signaling transduction.
As they are highly heterogeneous, tumors have distinct metabolic signatures, and identifying tumor-specific biomarkers has the potential to improve precise cancer diagnosis. Additionally, there is ample evidence to support the anti-tumor efficacy of targeting this metabolic vulnerability alone or in combination. Notably, metabolic pathways are intertwined and largely overlap. A complete blockade may cause active compensatory supply, impairing the inhibitory effects, highlighting the importance of monitoring metabolite dynamics and moderate intervention.
What we have described here is merely the tip of the iceberg, providing the impetus for further investigation. Studies are needed to uncover how metabolites affect the expression of specific genes and signaling pathways in more detail. Furthermore, metabolites exist both outside and inside different cellular compartments, and it would be of interest to explore how metabolite transportation and localization are regulated. It is likely that additional extra-metabolic functions of metabolites will be identified in the near future, which will have far-reaching implications for the understanding of tumor biology and improving translational clinical approaches.
Abbreviations
1,3-BPG, 1,3-bisphosphoglycerate; 2-HG, 2-hydroxyglutarate; 2-PG, 2-phosphoglycerate; 3-OHB, 3-hydroxybutyrate; 3-PG, 3-phosphoglycerate; 6-PG, 6-phosphogluconate; AcAc-CoA, acetoacetyl-CoA; Ac-CoA, acetyl-CoA; ACL, ATP citrate lyase; ACSS, ac-CoA synthetase; AhR, aryl hydrocarbon receptor; D2HGDH, D-2-hydroxyglutarate dehydrogenase; EMT, epithelial-to-mesenchymal transition; FABP5, fatty acid-binding protein 5; FH, fumarate hydratase; G6P, glucose-6-phosphate; G6PD, glucose-6-phosphate dehydrogenase; GA3P, glyceraldehyde-3-phosphate; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; GCL, glutamate cysteine ligase; GLS, glutaminases; GLUD, glutamate dehydrogenase; GPR, G-protein coupled receptors; GSH, glutathione; GSS, GSH synthetase; HIF1α, hypoxia-inducible factor 1-alpha; HMGCL, 3-hydroxy-3-methylglutaryl-CoA lyase; HMG-CoA, 3-hydroxy-3-methylglutaryl-CoA; HMGCS, 3-hydroxy-3-methylglutaryl–CoA synthase; ICL, isocitrate lyase; IDH, isocitrate dehydrogenase; Kbhb, lysine β-hydroxybutyrylation; KDHs, ketoacid dehydrogenases; L2HGDH, L-2-hydroxyglutarate dehydrogenase; KP, kynurenine (Kyn) pathway; KYNU, kynureninase; LCFA, long chain fatty acids; LDH, lactate dehydrogenase; LDHA, lactate dehydrogenase A; MAT2A, methionine adenosyltransferase 2A; MCT1, monocarboxylate transporters; MDH, malate dehydrogenase; MeS, metabolite-sensing module; MeSr, metabolite sensor-mediated signaling; MTs, methyltransferases; NF-κB, nuclear factor κB; PDH, pyruvate dehydrogenase; PEP, phosphoenolpyruvate; PGAM, phosphoglycerate mutase; PGD, phosphogluconate dehydrogenase; PGLS, 6-phosphogluconolactonase; PHD, prolyl hydroxylase domain; PHGDH, phosphoglycerate dehydrogenase; PPARβ/δ, peroxisome proliferator-activated receptor β/δ; PPP, pentose phosphate pathway; PTM, post-transcriptional modifications; R-5-P, ribose-5-phosphate; Ru-5-P, ribulose-5-phosphate; SAH, S-adenosyl-homocysteine; SAM, S-adenosyl-methionine; SC, sensing by conjugating; SDH, succinate dehydrogenase; TCA cycle, tricarboxylic acid cycle; δ-6-PGL, δ-6-phosphogluconolactone.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, have agreed on the journal to which the article will be submitted, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
- CairnsRA, HarrisIS, MakTW. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011;11(2):85–95. doi:10.1038/nrc298121258394
- LauAN, Vander HeidenMG. Metabolism in the tumor microenvironment. Ann Rev Cancer Biol. 2020;4(1):17–40. doi:10.1146/annurev-cancerbio-030419-033333
- PhanLM, YeungSC, LeeMH. Cancer metabolic reprogramming: importance, main features, and potentials for precise targeted anti-cancer therapies. Cancer Biol Med. 2014;11(1):1–19.24738035
- YoshidaGJ. Metabolic reprogramming: the emerging concept and associated therapeutic strategies. J Exp Clin Cancer Res. 2015;34(1):111. doi:10.1186/s13046-015-0221-y26445347
- SullivanLB, GuiDY, Vander HeidenMG. Altered metabolite levels in cancer: implications for tumour biology and cancer therapy. Nat Rev Cancer. 2016;16(11):680–693. doi:10.1038/nrc.2016.8527658530
- GilliesRJ, RobeyI, GatenbyRA. Causes and consequences of increased glucose metabolism of cancers. J Nucl Med. 2008;49(Suppl 2):24s–42s. doi:10.2967/jnumed.107.04725818523064
- KingA, SelakMA, GottliebE. Succinate dehydrogenase and fumarate hydratase: linking mitochondrial dysfunction and cancer. Oncogene. 2006;25(34):4675–4682. doi:10.1038/sj.onc.120959416892081
- WardPS, PatelJ, WiseDR, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17(3):225–234. doi:10.1016/j.ccr.2010.01.02020171147
- YangM, SogaT, PollardPJ. Oncometabolites: linking altered metabolism with cancer. J Clin Invest. 2013;123(9):3652–3658. doi:10.1172/JCI6722823999438
- NalbantogluS, KaradagA. Metabolomics bridging proteomics along metabolites/oncometabolites and protein modifications: paving the way toward integrative multiomics. J Pharm Biomed Anal. 2021;199:114031.33857836
- KinnairdA, ZhaoS, WellenKE, MichelakisED. Metabolic control of epigenetics in cancer. Nat Rev Cancer. 2016;16(11):694–707. doi:10.1038/nrc.2016.8227634449
- StramAR, PayneRM. Post-translational modifications in mitochondria: protein signaling in the powerhouse. Cell Mol Life Sci. 2016;73(21):4063–4073. doi:10.1007/s00018-016-2280-427233499
- WangYP, LeiQY. Metabolite sensing and signaling in cell metabolism. Signal Transduct Target Ther. 2018;3(1):30. doi:10.1038/s41392-018-0024-730416760
- AlfaroukKO, VerduzcoD, RauchC, et al. Glycolysis, tumor metabolism, cancer growth and dissemination. A new pH-based etiopathogenic perspective and therapeutic approach to an old cancer question. Oncoscience. 2014;1(12):777–802. doi:10.18632/oncoscience.10925621294
- MoelleringRE, CravattBF. Functional lysine modification by an intrinsically reactive primary glycolytic metabolite. Science. 2013;341(6145):549–553. doi:10.1126/science.123832723908237
- OslundRC, SuX, HaugbroM, et al. Bisphosphoglycerate mutase controls serine pathway flux via 3-phosphoglycerate. Nat Chem Biol. 2017;13(10):1081–1087. doi:10.1038/nchembio.245328805803
- HitosugiT, ZhouL, ElfS, et al. Phosphoglycerate mutase 1 coordinates glycolysis and biosynthesis to promote tumor growth. Cancer Cell. 2012;22(5):585–600. doi:10.1016/j.ccr.2012.09.02023153533
- HoPC, BihuniakJD, MacintyreAN, et al. Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell. 2015;162(6):1217–1228. doi:10.1016/j.cell.2015.08.01226321681
- Moreno-FeliciJ, HyroššováP, AragóM, et al. Phosphoenolpyruvate from glycolysis and PEPCK regulate cancer cell fate by altering cytosolic Ca(2). Cells. 2019;9(1):18. doi:10.3390/cells9010018
- San-MillánI, BrooksGA. Reexamining cancer metabolism: lactate production for carcinogenesis could be the purpose and explanation of the Warburg effect. Carcinogenesis. 2017;38(2):119–133.27993896
- IppolitoL, MorandiA, GiannoniE, ChiarugiP. Lactate: a metabolic driver in the tumour landscape. Trends Biochem Sci. 2019;44(2):153–166. doi:10.1016/j.tibs.2018.10.01130473428
- De SaedeleerCJ, CopettiT, PorporatoPE, VerraxJ, FeronO, SonveauxP. Lactate activates HIF-1 in oxidative but not in Warburg-phenotype human tumor cells. PLoS One. 2012;7(10):e46571. doi:10.1371/journal.pone.004657123082126
- VégranF, BoidotR, MichielsC, SonveauxP, FeronO. Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports an NF-κB/IL-8 pathway that drives tumor angiogenesis. Cancer Res. 2011;71(7):2550–2560. doi:10.1158/0008-5472.CAN-10-282821300765
- LeeDC, SohnHA, ParkZY, et al. A lactate-induced response to hypoxia. Cell. 2015;161(3):595–609. doi:10.1016/j.cell.2015.03.01125892225
- ColegioOR, ChuNQ, SzaboAL, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. 2014;513(7519):559–563. doi:10.1038/nature1349025043024
- KesMMG, Van den BosscheJ, GriffioenAW, HuijbersEJM. Oncometabolites lactate and succinate drive pro-angiogenic macrophage response in tumors. Biochim Biophys Acta Rev Cancer. 2020;1874(2):188427. doi:10.1016/j.bbcan.2020.18842732961257
- MuX, ShiW, XuY, et al. Tumor-derived lactate induces M2 macrophage polarization via the activation of the ERK/STAT3 signaling pathway in breast cancer. Cell Cycle. 2018;17(4):428–438. doi:10.1080/15384101.2018.144430529468929
- RolandCL, ArumugamT, DengD, et al. Cell surface lactate receptor GPR81 is crucial for cancer cell survival. Cancer Res. 2014;74(18):5301–5310. doi:10.1158/0008-5472.CAN-14-031924928781
- FengJ, YangH, ZhangY, et al. Tumor cell-derived lactate induces TAZ-dependent upregulation of PD-L1 through GPR81 in human lung cancer cells. Oncogene. 2017;36(42):5829–5839. doi:10.1038/onc.2017.18828604752
- BrownTP, GanapathyV. Lactate/GPR81 signaling and proton motive force in cancer: role in angiogenesis, immune escape, nutrition, and Warburg phenomenon. Pharmacol Ther. 2020;206:107451. doi:10.1016/j.pharmthera.2019.10745131836453
- YangK, XuJ, FanM, et al. Lactate suppresses macrophage pro-inflammatory response to LPS stimulation by inhibition of YAP and NF-κB activation via GPR81-mediated signaling. Front Immunol. 2020;11:587913. doi:10.3389/fimmu.2020.58791333123172
- ZhangW, WangG, XuZG, et al. Lactate is a natural suppressor of RLR signaling by targeting MAVS. Cell. 2019;178(1):176–189.e115. doi:10.1016/j.cell.2019.05.00331155231
- LathamT, MackayL, SproulD, et al. Lactate, a product of glycolytic metabolism, inhibits histone deacetylase activity and promotes changes in gene expression. Nucleic Acids Res. 2012;40(11):4794–4803. doi:10.1093/nar/gks06622323521
- ZhangD, TangZ, HuangH, et al. Metabolic regulation of gene expression by histone lactylation. Nature. 2019;574(7779):575–580. doi:10.1038/s41586-019-1678-131645732
- PatraKC, HayN. The pentose phosphate pathway and cancer. Trends Biochem Sci. 2014;39(8):347–354. doi:10.1016/j.tibs.2014.06.00525037503
- ShackelfordDB, ShawRJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. 2009;9(8):563–575. doi:10.1038/nrc267619629071
- ParkS, SchefflerTL, RossieSS, GerrardDE. AMPK activity is regulated by calcium-mediated protein phosphatase 2A activity. Cell Calcium. 2013;53(3):217–223. doi:10.1016/j.ceca.2012.12.00123298795
- LinR, ElfS, ShanC, et al. 6-Phosphogluconate dehydrogenase links oxidative PPP, lipogenesis and tumour growth by inhibiting LKB1-AMPK signalling. Nat Cell Biol. 2015;17(11):1484–1496. doi:10.1038/ncb325526479318
- GaoX, ZhaoL, LiuS, et al. γ-6-Phosphogluconolactone, a byproduct of the oxidative pentose phosphate pathway, contributes to AMPK activation through inhibition of PP2A. Mol Cell. 2019;76(6):857–871.e859. doi:10.1016/j.molcel.2019.09.00731586547
- AndersonNM, MuckaP, KernJG, FengH. The emerging role and targetability of the TCA cycle in cancer metabolism. Protein Cell. 2018;9(2):216–237. doi:10.1007/s13238-017-0451-128748451
- DangL, WhiteDW, GrossS, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462(7274):739–744. doi:10.1038/nature0861719935646
- YanH, ParsonsDW, JinG, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360(8):765–773. doi:10.1056/NEJMoa080871019228619
- MardisER, DingL, DoolingDJ, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361(11):1058–1066. doi:10.1056/NEJMoa090384019657110
- ParsonsDW, JonesS, ZhangX, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321(5897):1807–1812. doi:10.1126/science.116438218772396
- YangH, YeD, GuanKL, XiongY. IDH1 and IDH2 mutations in tumorigenesis: mechanistic insights and clinical perspectives. Clin Cancer Res. 2012;18(20):5562–5571. doi:10.1158/1078-0432.CCR-12-177323071358
- DangL, SuSM. Isocitrate dehydrogenase mutation and (R)-2-hydroxyglutarate: from basic discovery to therapeutics development. Annu Rev Biochem. 2017;86(1):305–331. doi:10.1146/annurev-biochem-061516-04473228375741
- FanJ, TengX, LiuL, et al. Human phosphoglycerate dehydrogenase produces the oncometabolite D-2-hydroxyglutarate. ACS Chem Biol. 2015;10(2):510–516. doi:10.1021/cb500683c25406093
- RzemR, VincentMF, Van SchaftingenE, Veiga-da-cunhaM. L-2-hydroxyglutaric aciduria, a defect of metabolite repair. J Inherit Metab Dis. 2007;30(5):681–689. doi:10.1007/s10545-007-0487-017603759
- IntlekoferAM, DematteoRG, VennetiS, et al. Hypoxia induces production of L-2-hydroxyglutarate. Cell Metab. 2015;22(2):304–311.26212717
- StruysEA, SalomonsGS, AchouriY, et al. Mutations in the D-2-hydroxyglutarate dehydrogenase gene cause D-2-hydroxyglutaric aciduria. Am J Hum Genet. 2005;76(2):358–360. doi:10.1086/42789015609246
- AchouriY, NoëlG, VertommenD, RiderMH, Veiga-da-cunhaM, Van SchaftingenE. Identification of a dehydrogenase acting on D-2-hydroxyglutarate. Biochem J. 2004;381(Pt 1):35–42. doi:10.1042/BJ2003193315070399
- RzemR, Veiga-da-cunhaM, NoëlG, et al. A gene encoding a putative FAD-dependent L-2-hydroxyglutarate dehydrogenase is mutated in L-2-hydroxyglutaric aciduria. Proc Natl Acad Sci U S A. 2004;101(48):16849–16854. doi:10.1073/pnas.040484010115548604
- XuW, YangH, LiuY, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19(1):17–30. doi:10.1016/j.ccr.2010.12.01421251613
- ChowdhuryR, YeohKK, TianYM, et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011;12(5):463–469. doi:10.1038/embor.2011.4321460794
- LosmanJA, KoivunenP, KaelinWGJr. 2-Oxoglutarate-dependent dioxygenases in cancer. Nat Rev Cancer. 2020;20(12):710–726.33087883
- ShiY, WhetstineJR. Dynamic regulation of histone lysine methylation by demethylases. Mol Cell. 2007;25(1):1–14. doi:10.1016/j.molcel.2006.12.01017218267
- LuC, WardPS, KapoorGS, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483(7390):474–478. doi:10.1038/nature1086022343901
- CarbonneauM, LalondeL, LalondeM-E. The oncometabolite 2-hydroxyglutarate activates the mTOR signalling pathway. Nat Commun. 2016;7(1):12700. doi:10.1038/ncomms1270027624942
- TahilianiM, KohKP, ShenY, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324(5929):930–935. doi:10.1126/science.117011619372391
- FigueroaME, Abdel-WahabO, LuC, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18(6):553–567. doi:10.1016/j.ccr.2010.11.01521130701
- ZhaoS, LinY, XuW, et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science. 2009;324(5924):261–265. doi:10.1126/science.117094419359588
- TarhonskayaH, RydzikAM, LeungIK, et al. Non-enzymatic chemistry enables 2-hydroxyglutarate-mediated activation of 2-oxoglutarate oxygenases. Nat Commun. 2014;5(1):3423. doi:10.1038/ncomms442324594748
- KeithB, JohnsonRS, SimonMC. HIF1α and HIF2α: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer. 2011;12(1):9–22. doi:10.1038/nrc318322169972
- WangP, WuJ, MaS, et al. Oncometabolite D-2-hydroxyglutarate inhibits ALKBH DNA repair enzymes and sensitizes IDH mutant cells to alkylating agents. Cell Rep. 2015;13(11):2353–2361. doi:10.1016/j.celrep.2015.11.02926686626
- GillAJ. Succinate dehydrogenase (SDH)-deficient neoplasia. Histopathology. 2018;72(1):106–116. doi:10.1111/his.1327729239034
- ZhaoT, MuX, YouQ. Succinate: an initiator in tumorigenesis and progression. Oncotarget. 2017;8(32):53819–53828. doi:10.18632/oncotarget.1773428881853
- RasolaA, NeckersL, PicardD. Mitochondrial oxidative phosphorylation TRAP(1)ped in tumor cells. Trends Cell Biol. 2014;24(8):455–463. doi:10.1016/j.tcb.2014.03.00524731398
- XiaoM, YangH, XuW, et al. Inhibition of α-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012;26(12):1326–1338. doi:10.1101/gad.191056.11222677546
- LetouzéE, MartinelliC, LoriotC, et al. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell. 2013;23(6):739–752. doi:10.1016/j.ccr.2013.04.01823707781
- LoriotC, BurnichonN, GadessaudN, et al. Epithelial to mesenchymal transition is activated in metastatic pheochromocytomas and paragangliomas caused by SDHB gene mutations. J Clin Endocrinol Metab. 2012;97(6):E954–962. doi:10.1210/jc.2011-343722492777
- LoriotC, DominguesM, BergerA, et al. Deciphering the molecular basis of invasiveness in Sdhb-deficient cells. Oncotarget. 2015;6(32):32955–32965. doi:10.18632/oncotarget.510626460615
- RapizziE, ErcolinoT, FucciR, et al. Succinate dehydrogenase subunit B mutations modify human neuroblastoma cell metabolism and proliferation. Horm Cancer. 2014;5(3):174–184. doi:10.1007/s12672-014-0172-324595825
- HoekstraAS, de GraaffMA, Briaire-de BruijnIH, et al. Inactivation of SDH and FH cause loss of 5hmC and increased H3K9me3 in paraganglioma/pheochromocytoma and smooth muscle tumors. Oncotarget. 2015;6(36):38777–38788. doi:10.18632/oncotarget.609126472283
- SelakMA, ArmourSM, MacKenzieED, et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell. 2005;7(1):77–85. doi:10.1016/j.ccr.2004.11.02215652751
- TretterL, PatocsA, ChinopoulosC. Succinate, an intermediate in metabolism, signal transduction, ROS, hypoxia, and tumorigenesis. Biochim Biophys Acta. 2016;1857(8):1086–1101. doi:10.1016/j.bbabio.2016.03.01226971832
- SmestadJ, ErberL, ChenY, MaherLJ3rd. Chromatin succinylation correlates with active gene expression and is perturbed by defective TCA cycle metabolism. iScience. 2018;2:63–75. doi:10.1016/j.isci.2018.03.01229888767
- Dalla PozzaE, DandoI, PacchianaR, et al. Regulation of succinate dehydrogenase and role of succinate in cancer. Semin Cell Dev Biol. 2020;98:4–14. doi:10.1016/j.semcdb.2019.04.01331039394
- LiuC, LiuY, ChenL, et al. Quantitative proteome and lysine succinylome analyses provide insights into metabolic regulation in breast cancer. Breast Cancer. 2019;26(1):93–105. doi:10.1007/s12282-018-0893-130022435
- LiX, ZhangC, ZhaoT, et al. Lysine-222 succinylation reduces lysosomal degradation of lactate dehydrogenase a and is increased in gastric cancer. J Exp Clin Cancer Res. 2020;39(1):172. doi:10.1186/s13046-020-01681-032859246
- MuX, ZhaoT, XuC, et al. Oncometabolite succinate promotes angiogenesis by upregulating VEGF expression through GPR91-mediated STAT3 and ERK activation. Oncotarget. 2017;8(8):13174–13185. doi:10.18632/oncotarget.1448528061458
- WuJY, HuangTW, HsiehYT, et al. Cancer-derived succinate promotes macrophage polarization and cancer metastasis via succinate receptor. Mol Cell. 2020;77(2):213–227.e215. doi:10.1016/j.molcel.2019.10.02331735641
- TomlinsonIP, AlamNA, RowanAJ, et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet. 2002;30(4):406–410.11865300
- BardellaC, El-BahrawyM, FrizzellN, et al. Aberrant succination of proteins in fumarate hydratase-deficient mice and HLRCC patients is a robust biomarker of mutation status. J Pathol. 2011;225(1):4–11. doi:10.1002/path.293221630274
- BlatnikM, FrizzellN, ThorpeSR, BaynesJW. Inactivation of glyceraldehyde-3-phosphate dehydrogenase by fumarate in diabetes: formation of S-(2-succinyl)cysteine, a novel chemical modification of protein and possible biomarker of mitochondrial stress. Diabetes. 2008;57(1):41–49. doi:10.2337/db07-083817934141
- FrizzellN, RajeshM, JepsonMJ, et al. Succination of thiol groups in adipose tissue proteins in diabetes: succination inhibits polymerization and secretion of adiponectin. J Biol Chem. 2009;284(38):25772–25781. doi:10.1074/jbc.M109.01925719592500
- KinchL, GrishinNV, BrugarolasJ. Succination of Keap1 and activation of Nrf2-dependent antioxidant pathways in FH-deficient papillary renal cell carcinoma type 2. Cancer Cell. 2011;20(4):418–420. doi:10.1016/j.ccr.2011.10.00522014567
- AdamJ, HatipogluE, O’FlahertyL, et al. Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell. 2011;20(4):524–537. doi:10.1016/j.ccr.2011.09.00622014577
- SullivanLB, Martinez-GarciaE, NguyenH, et al. The proto-oncometabolite fumarate binds glutathione to amplify ROS-dependent signaling. Mol Cell. 2013;51(2):236–248. doi:10.1016/j.molcel.2013.05.00323747014
- ZhengL, CardaciS, JerbyL, et al. Fumarate induces redox-dependent senescence by modifying glutathione metabolism. Nat Commun. 2015;6(1):6001. doi:10.1038/ncomms700125613188
- YoshiiY, FurukawaT, SagaT, FujibayashiY. Acetate/acetyl-CoA metabolism associated with cancer fatty acid synthesis: overview and application. Cancer Lett. 2015;356(2Pt A):211–216. doi:10.1016/j.canlet.2014.02.01924569091
- YoshiiY, WakiA, FurukawaT, et al. Tumor uptake of radiolabeled acetate reflects the expression of cytosolic acetyl-CoA synthetase: implications for the mechanism of acetate PET. Nucl Med Biol. 2009;36(7):771–777. doi:10.1016/j.nucmedbio.2009.05.00619720289
- BauerDE, HatzivassiliouG, ZhaoF, AndreadisC, ThompsonCB. ATP citrate lyase is an important component of cell growth and transformation. Oncogene. 2005;24(41):6314–6322. doi:10.1038/sj.onc.120877316007201
- SchugZT, PeckB, JonesDT, et al. Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell. 2015;27(1):57–71. doi:10.1016/j.ccell.2014.12.00225584894
- MetalloCM, GameiroPA, BellEL, et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature. 2011;481(7381):380–384. doi:10.1038/nature1060222101433
- LeeJV, CarrerA, ShahS, et al. Akt-dependent metabolic reprogramming regulates tumor cell histone acetylation. Cell Metab. 2014;20(2):306–319. doi:10.1016/j.cmet.2014.06.00424998913
- TrefelyS, LovellCD, SnyderNW, WellenKE. Compartmentalised acyl-CoA metabolism and roles in chromatin regulation. Mol Metab. 2020;38:100941. doi:10.1016/j.molmet.2020.01.00532199817
- McDonnellE, CrownSB, FoxDB, et al. Lipids reprogram metabolism to become a major carbon source for histone acetylation. Cell Rep. 2016;17(6):1463–1472. doi:10.1016/j.celrep.2016.10.01227806287
- CarrerA, TrefelyS, ZhaoS, et al. Acetyl-CoA metabolism supports multistep pancreatic tumorigenesis. Cancer Discov. 2019;9(3):416–435. doi:10.1158/2159-8290.CD-18-056730626590
- ZhengZQ, LiZX, GuanJL, et al. Long noncoding RNA TINCR-mediated regulation of acetyl-CoA metabolism promotes nasopharyngeal carcinoma progression and chemoresistance. Cancer Res. 2020;80(23):5174–5188. doi:10.1158/0008-5472.CAN-19-362633067266
- GaoX, LinSH, RenF, et al. Acetate functions as an epigenetic metabolite to promote lipid synthesis under hypoxia. Nat Commun. 2016;7(1):11960. doi:10.1038/ncomms1196027357947
- LinR, TaoR, GaoX, et al. Acetylation stabilizes ATP-citrate lyase to promote lipid biosynthesis and tumor growth. Mol Cell. 2013;51(4):506–518. doi:10.1016/j.molcel.2013.07.00223932781
- ChenD, XiaS, ZhangR, et al. Lysine acetylation restricts mutant IDH2 activity to optimize transformation in AML cells. Mol Cell. 2021;S1097-2765(21)00507-4. doi:10.1016/j.molcel.2021.06.027
- BoseS, RameshV, LocasaleJW. Acetate metabolism in physiology, cancer, and beyond. Trends Cell Biol. 2019;29(9):695–703. doi:10.1016/j.tcb.2019.05.00531160120
- SchugZT, Vande VoordeJ, GottliebE. The metabolic fate of acetate in cancer. Nat Rev Cancer. 2016;16(11):708–717. doi:10.1038/nrc.2016.8727562461
- HatanakaH, TsukuiM, TakadaS, et al. Identification of transforming activity of free fatty acid receptor 2 by retroviral expression screening. Cancer Sci. 2010;101(1):54–59. doi:10.1111/j.1349-7006.2009.01348.x19780758
- YonezawaT, KobayashiY, ObaraY. Short-chain fatty acids induce acute phosphorylation of the p38 mitogen-activated protein kinase/heat shock protein 27 pathway via GPR43 in the MCF-7 human breast cancer cell line. Cell Signal. 2007;19(1):185–193. doi:10.1016/j.cellsig.2006.06.00416887331
- LeviL, WangZ, DoudMK, HazenSL, NoyN. Saturated fatty acids regulate retinoic acid signalling and suppress tumorigenesis by targeting fatty acid-binding protein 5. Nat Commun. 2015;6(1):8794. doi:10.1038/ncomms979426592976
- ArmstrongEH, GoswamiD, GriffinPR, NoyN, OrtlundEA. Structural basis for ligand regulation of the fatty acid-binding protein 5, peroxisome proliferator-activated receptor β/δ (FABP5-PPARβ/δ) signaling pathway. J Biol Chem. 2014;289(21):14941–14954. doi:10.1074/jbc.M113.51464624692551
- LongoR, PeriC, CricrìD, et al. Ketogenic diet: a new light shining on old but gold biochemistry. Nutrients. 2019;11(10):2497. doi:10.3390/nu11102497
- AbdelmegeedMA, KimSK, WoodcroftKJ, NovakRF. Acetoacetate activation of extracellular signal-regulated kinase 1/2 and p38 mitogen-activated protein kinase in primary cultured rat hepatocytes: role of oxidative stress. J Pharmacol Exp Ther. 2004;310(2):728–736. doi:10.1124/jpet.104.06652215051799
- KangHB, FanJ, LinR, et al. Metabolic rewiring by oncogenic BRAF V600E links ketogenesis pathway to BRAF-MEK1 signaling. Mol Cell. 2015;59(3):345–358. doi:10.1016/j.molcel.2015.05.03726145173
- ZhaoL, FanJ, XiaS, et al. HMG-CoA synthase 1 is a synthetic lethal partner of BRAF(V600E) in human cancers. J Biol Chem. 2017;292(24):10142–10152. doi:10.1074/jbc.M117.78877828468827
- XiaS, LinR, JinL, et al. Prevention of dietary-fat-fueled ketogenesis attenuates BRAF V600E tumor growth. Cell Metab. 2017;25(2):358–373. doi:10.1016/j.cmet.2016.12.01028089569
- NewmanJC, VerdinE. β-hydroxybutyrate: much more than a metabolite. Diabetes Res Clin Pract. 2014;106(2):173–181. doi:10.1016/j.diabres.2014.08.00925193333
- NewmanJC, VerdinE. Ketone bodies as signaling metabolites. Trends Endocrinol Metab. 2014;25(1):42–52. doi:10.1016/j.tem.2013.09.00224140022
- MollerN. Ketone body, 3-hydroxybutyrate: minor metabolite - major medical manifestations. J Clin Endocrinol Metab. 2020;105(9):2884–2892. doi:10.1210/clinem/dgaa370
- DabekA, WojtalaM, PirolaL, BalcerczykA. Modulation of cellular biochemistry, epigenetics and metabolomics by ketone bodies. Implications of the ketogenic diet in the physiology of the organism and pathological states. Nutrients. 2020;12(3):788. doi:10.3390/nu12030788
- ShimazuT, HirscheyMD, NewmanJ, et al. Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science. 2013;339(6116):211–214. doi:10.1126/science.122716623223453
- XieZ, ZhangD, ChungD, et al. Metabolic regulation of gene expression by histone lysine β-hydroxybutyrylation. Mol Cell. 2016;62(2):194–206. doi:10.1016/j.molcel.2016.03.03627105115
- LiuK, LiF, SunQ, et al. p53 β-hydroxybutyrylation attenuates p53 activity. Cell Death Dis. 2019;10(3):243. doi:10.1038/s41419-019-1463-y30858356
- RisticB, BhutiaYD, GanapathyV. Cell-surface G-protein-coupled receptors for tumor-associated metabolites: a direct link to mitochondrial dysfunction in cancer. Biochim Biophys Acta Rev Cancer. 2017;1868(1):246–257. doi:10.1016/j.bbcan.2017.05.00328512002
- SandersonSM, GaoX, DaiZ, LocasaleJW. Methionine metabolism in health and cancer: a nexus of diet and precision medicine. Nat Rev Cancer. 2019;19(11):625–637. doi:10.1038/s41568-019-0187-831515518
- SchmidtT, LehaA, Salinas-RiesterG. Treatment of prostate cancer cells with S-adenosylmethionine leads to genome-wide alterations in transcription profiles. Gene. 2016;595(2):161–167. doi:10.1016/j.gene.2016.09.03227688072
- MentchSJ, MehrmohamadiM, HuangL, et al. Histone methylation dynamics and gene regulation occur through the sensing of one-carbon metabolism. Cell Metab. 2015;22(5):861–873. doi:10.1016/j.cmet.2015.08.02426411344
- ShukeirN, PakneshanP, ChenG, SzyfM, RabbaniSA. Alteration of the methylation status of tumor-promoting genes decreases prostate cancer cell invasiveness and tumorigenesis in vitro and in vivo. Cancer Res. 2006;66(18):9202–9210. doi:10.1158/0008-5472.CAN-06-195416982764
- ChikF, MachnesZ, SzyfM. Synergistic anti-breast cancer effect of a combined treatment with the methyl donor S-adenosyl methionine and the DNA methylation inhibitor 5-aza-2ʹ-deoxycytidine. Carcinogenesis. 2014;35(1):138–144. doi:10.1093/carcin/bgt28423985780
- LuoJ, LiYN, WangF, ZhangWM, GengX. S-adenosylmethionine inhibits the growth of cancer cells by reversing the hypomethylation status of c-myc and H-ras in human gastric cancer and colon cancer. Int J Biol Sci. 2010;6(7):784–795. doi:10.7150/ijbs.6.78421152119
- LiTW, YangH, PengH, XiaM, MatoJM, LuSC. Effects of S-adenosylmethionine and methylthioadenosine on inflammation-induced colon cancer in mice. Carcinogenesis. 2012;33(2):427–435. doi:10.1093/carcin/bgr29522159228
- LiTW, PengH, YangH, et al. S-Adenosylmethionine and methylthioadenosine inhibit β-catenin signaling by multiple mechanisms in liver and colon cancer. Mol Pharmacol. 2015;87(1):77–86. doi:10.1124/mol.114.09567925338671
- IlissoCP, SapioL, Delle CaveD, et al. S-Adenosylmethionine affects ERK1/2 and Stat3 pathways and induces apotosis in osteosarcoma cells. J Cell Physiol. 2016;231(2):428–435. doi:10.1002/jcp.2508926174106
- AltmanBJ, StineZE, DangCV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer. 2016;16(10):619–634. doi:10.1038/nrc.2016.7127492215
- BansalA, SimonMC. Glutathione metabolism in cancer progression and treatment resistance. J Cell Biol. 2018;217(7):2291–2298. doi:10.1083/jcb.20180416129915025
- SinghS, KhanAR, GuptaAK. Role of glutathione in cancer pathophysiology and therapeutic interventions. J Exp Ther Oncol. 2012;9(4):303–316.22545423
- ZhangJ, YeZW, SinghS, TownsendDM, TewKD. An evolving understanding of the S-glutathionylation cycle in pathways of redox regulation. Free Radic Biol Med. 2018;120:204–216. doi:10.1016/j.freeradbiomed.2018.03.03829578070
- TewKD, ManevichY, GrekC, XiongY, UysJ, TownsendDM. The role of glutathione S-transferase P in signaling pathways and S-glutathionylation in cancer. Free Radic Biol Med. 2011;51(2):299–313. doi:10.1016/j.freeradbiomed.2011.04.01321558000
- García-GiménezJL, ÒlasoG, HakeSB, et al. Histone h3 glutathionylation in proliferating mammalian cells destabilizes nucleosomal structure. Antioxid Redox Signal. 2013;19(12):1305–1320. doi:10.1089/ars.2012.502123541030
- PlattenM, NollenEAA, RohrigUF, FallarinoF, OpitzCA. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat Rev Drug Discov. 2019;18(5):379–401.30760888
- LiuXH, ZhaiXY. Role of tryptophan metabolism in cancers and therapeutic implications. Biochimie. 2021;182:131–139. doi:10.1016/j.biochi.2021.01.00533460767
- WalczakK, LangnerE, Makuch-KockaA, et al. Effect of tryptophan-derived ahr ligands, kynurenine, kynurenic acid and FICZ, on proliferation, cell cycle regulation and cell death of melanoma cells-in vitro studies. Int J Mol Sci. 2020;21(21):7946. doi:10.3390/ijms21217946
- KolluriSK, JinUH, SafeS. Role of the aryl hydrocarbon receptor in carcinogenesis and potential as an anti-cancer drug target. Arch Toxicol. 2017;91(7):2497–2513. doi:10.1007/s00204-017-1981-228508231
- VenkateswaranN, Lafita-NavarroMC, HaoYH, et al. MYC promotes tryptophan uptake and metabolism by the kynurenine pathway in colon cancer. Genes Dev. 2019;33(17–18):1236–1251. doi:10.1101/gad.327056.11931416966
- CampesatoLF, BudhuS, TchaichaJ, et al. Blockade of the AHR restricts a Treg-macrophage suppressive axis induced by L-Kynurenine. Nat Commun. 2020;11(1):4011. doi:10.1038/s41467-020-17750-z32782249
- DiNataleBC, MurrayIA, SchroederJC, et al. Kynurenic acid is a potent endogenous aryl hydrocarbon receptor ligand that synergistically induces interleukin-6 in the presence of inflammatory signaling. Toxicol Sci. 2010;115(1):89–97. doi:10.1093/toxsci/kfq02420106948
- WalczakK, WnorowskiA, TurskiWA, PlechT. Kynurenic acid and cancer: facts and controversies. Cell Mol Life Sci. 2020;77(8):1531–1550. doi:10.1007/s00018-019-03332-w31659416
- WalczakK, Deneka-HannemannS, JaroszB, et al. Kynurenic acid inhibits proliferation and migration of human glioblastoma T98G cells. Pharmacol Rep. 2014;66(1):130–136. doi:10.1016/j.pharep.2013.06.00724905318