
Abstract
Mature overall survival (OS) data are often unavailable at the time of regulatory and reimbursement decisions for a new cancer treatment. For patients with early-stage cancers treated with potentially curative treatments, demonstrating an OS benefit may take years and may be confounded by subsequent lines of therapy or crossover to the investigational treatment. For patients with advanced-stage cancers, mature OS data may be available but difficult to interpret for similar reasons. There are strong opinions about approval and reimbursement in the absence of mature OS data, with concerns over delay in patient access set against concerns about uncertainty in long-term benefit. This position paper reflects our individual views as patient advocate, clinician or health economist on one aspect of this debate. We look at payer decisions in the absence of mature OS data, considering when and how non-OS trial outcomes could inform decision-making and how uncertainty can be addressed beyond the trial, supporting these views with evidence from the literature. We consider when it is reasonable for payers to expect or not expect mature OS data at the initial reimbursement decision (based on criteria such as cancer stage and treatment efficacy) acknowledging that there are settings in which mature OS data are expected. We propose flexible strategies for generating and appraising patient-relevant evidence, including context-relevant endpoints and quality of life measures, when survival rates are good and mature OS data are not expected. We note that fair reimbursement is important; this means valuing patient benefit as shown through prespecified endpoints and reappraising if there is ongoing uncertainty or failure to show a sustained benefit. We suggest that reimbursement systems continue to evolve to align with scientific advances, because innovation is only meaningful if readily accessible to patients. The proposed strategies have the potential to promote thorough assessment of potential benefit to patients and lead to timely access to effective medicines.
Graphical Abstract
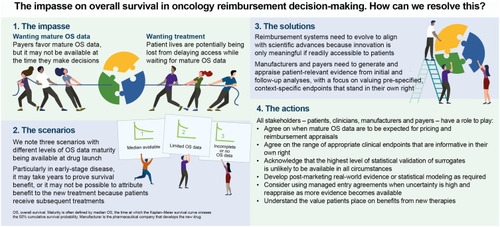
Plain Language Summary
Before patients can receive a new drug, regulators decide if it is safe and effective in treating the disease. Payers then decide if they will pay for the drug and, if so, at what price.
For cancer drugs, payers prefer to make decisions based on overall survival (OS) results. This is a measure of how long patients live after starting treatment. However, it may take many years to collect OS data. For example, patients with early-stage cancers may live for a long time with effective treatment. In addition, benefit can be difficult to measure if patients subsequently receive numerous other therapies.
If payers wait for long-term OS data, patients have to wait to access the new treatment, even when regulators have decided that it is safe and effective. As a result, some patients may die while waiting. On the other hand, if payers do not have long-term OS data, they risk paying for a treatment that is not as effective as they had hoped.
We use our individual views as a patient advocate, clinician or health economist to suggest how payers, clinicians, manufacturers and patients can work together to help patients access potentially life-extending drugs and payers spend money wisely.
We recommend using a range of clinical measures which can be measured earlier than OS, including delayed disease progression and improved quality of life.
We propose using methods to manage uncertainty, for example agreements between payers and pharmaceutical companies that are based on patient outcomes.
We suggest that payers evolve their decision-making so it aligns with advances in science. Innovation in new treatments is only meaningful if readily accessible to patients.
Introduction
Extending overall survival (OS) is an important treatment goal for many therapies in oncology. In clinical trials, OS is widely considered as the endpoint that is most valued by patients, clinicians, regulators and payers (see for common definitions). However, mature data on OS (eg, the median) are often not available at the time when reimbursement authorization decisions for new therapies are being made. In early-stage disease, when survival times are longer, proving an OS benefit can take a long time or may not be possible at all as therapies become more effective. In other cases, when cancer is advanced and a cure is unlikely, many patients will not survive long and OS data may be available relatively rapidly. However, even in late-stage disease, it may be difficult to interpret these data because it is hard to separate out the effect of a new treatment on OS when multiple subsequent lines of therapy have been used, or when patients on standard of care cross over to the new treatment either during or after the trial.Citation1 A tension then emerges between wanting to know whether, and by how much, a new treatment impacts on OS and needing to wait, sometimes for years, to obtain this information; or possibly never knowing with certainty. This tension is particularly acute for payers; given their primary objective of achieving the most health gain from their available budget, they need to be confident that the new treatment is providing value in terms of length of life, quality of life (QoL) or, ideally, both. There are strong opinions about approval and reimbursement in the absence of mature OS data, reflecting the high stakes at play, with concerns over delay in patient access set against concerns about the potential for treatments not to fully provide this anticipated benefit after longer follow-up and mature data.Citation2,Citation3 As payers have accountability to weigh the patient health benefit of new medicines against budget requirements, we see differences in expectations for OS among payers, and between payers and other stakeholders.Citation4–Citation6 In some cases, we see that regulators, clinicians and patients place different values on non-OS endpoints from payers.Citation3,Citation7,Citation8 This can lead to delayed access to regulatory-approved novel treatments, and in some cases eligible patients could die while these decisions are being made.Citation9 Time to reimbursement can differ significantly across health systemsCitation10 and some countries have implemented schemes to shorten the time to access;Citation11 however, schemes cover only some geographic regions and selected patients and are often not directly related to how payers appraise endpoints beyond OS.
Box 1 Definitions
Addressing and resolving the “OS impasse” is now more important than ever. Science is moving ahead quickly: our understanding of the complex molecular biology underlying different cancers is growing, and innovative, often personalized, new treatments are being developed. These advances, combined with earlier diagnosis and treatment, mean that patients are living longer with cancer or following cancer and it is increasingly difficult to present mature OS data at regulatory approval. Payer approval still, however, remains a requirement for translating innovation into better outcomes for patients and healthcare systems.Citation12
We have developed this position paper, taking our individual views gained from our experience as a patient advocate, clinician or health economist supported by evidence from the literature, and we focus on one aspect of this “OS impasse”. We look at payer decision-making in the absence of mature OS data, considering when and how non-OS trial outcomes could inform decisions and how uncertainty can be addressed beyond the trial. Our aim is collaborative, to work alongside payers and other stakeholders to reach a consensus on when it is appropriate to consider alternative endpoints in place of OS if mature OS data from clinical trials are not reasonably available; this means ensuring that evidence is relevant and that the uncertainty and risks faced by patients, clinicians, payers and manufacturers is mitigated. Our focus is evidence and endpoints, and our aim is for these considerations to apply across appraisal systems, whether based on cost-effectiveness or clinical/comparative effectiveness. While acknowledging the importance of drug pricing in decision-making and the trends in healthcare expenditure on cancer treatments,Citation13 we do not address these aspects in this paper.
Decisions are Increasingly Being Made Without Mature OS Data
Many regulatory and payer agencies consider OS as the most reliable and preferred endpoint ().Citation14,Citation19 Regulators acknowledge that the disease setting determines how endpoints can be considered for approval – the US Food and Drug Administration (FDA) notes that endpoint selection is highly dependent upon factors such as effect size and duration, depth of response, available therapy, location of disease, and the consequences of delaying or preventing disease progression or delaying use of subsequent toxic therapies.Citation14 These considerations are arguably broader than those employed by payers, and regulatory decisions are made without mature OS data for the majority of FDA and European Medicines Agency (EMA) oncology approvals. More than 70% of FDA adult cancer drug approvals between 2006 and 2017 were based on progression-free survival (PFS) and relapse-free survival.Citation6 Similarly, an analysis of EMA approvals for oncology treatments between 2014 and 2017 showed that in 34 of 88 (39%) marketing authorization applications, OS data were immature at the time of approval.Citation20 Payer guidelines have stated a clear preference for mature OS data when appraising cancer drugs for reimbursement (). Across cancer types, payer organizations (eg, Germany’s Federal Joint Committee [G-BA], the Spanish Agency of Medicines and Medical Products [AEMPS] and England’s National Institute for Health and Care Excellence [NICE])Citation21–Citation26 have critiqued the absence of mature OS data when appraising new therapies, in some instances denying, restricting or delaying patient access; in other instances, providing access despite criticizing the evidence.Citation27,Citation28
Table 1 Sample of Regulators’, Payers’ and Clinical Societies’ Positions on Types of Endpoints
Consequences of Decision-Making in the Absence of Mature OS Data
When improvement in OS is an important goal of therapy, decisions made in the absence of mature OS data come with risk and uncertainty, which can take different forms for different stakeholders. When the decision leads to no or delayed access, some patients can die waiting for OS to be demonstrated (see for an example), while others can lose the option to benefit from potentially life-extending future treatment (loss of option value).Citation39,Citation40 This can lead to patients, clinicians and payers accepting methods to manage uncertainty. Both the American Society of Clinical Oncology (ASCO) and the European Society for Medical Oncology (ESMO) have developed tools to help assess the relative benefit of cancer therapies (see ). However, these frameworks do not directly address how to enable patient access without mature OS data, in part because they are not regularly used by healthcare decision-makers, but also because of the focus on trial data rather than evidence beyond trials. Several countries have implemented schemes to shorten the time to access, such as reimbursement before the pricing decision (eg, in Germany) or early access (eg, Temporary Use Authorizations in France); elsewhere, coverage may be gained rapidly following regulatory approval (eg, in the USA and Japan).Citation11 Some countries have schemes to address uncertainty specifically around cancer drugs (eg, Cancer Drugs Fund in the UK). However, we note that these schemes cover only some geographic regions and selected patients, and are not directly related to how payers appraise endpoints beyond OS.
Box 2 Example for Impact of Earlier Time to Reimbursement for Patients
Although there have been advances in supporting patients in accessing new cancer medicines, we must acknowledge that early access comes with the risk of patients experiencing adverse events or inconvenience for no meaningful gain in length or QoL. Some of these risks can be mitigated through rescinding approval for drugs that do not show sustained patient benefit. This was seen in early 2021 when approval was withdrawn across a limited number of indications granted through the FDA Accelerated Approval Program, for which the drugs had not met post-marketing requirements.Citation41,Citation42
There are also financial consequences to decision-making. A consequence of reimbursement when value is not established is the use of healthcare budget that could have been used on more effective therapies.Citation2,Citation43,Citation44 A further financial consequence of delayed access is the lower than expected revenue for the manufacturer; this is felt immediately by the manufacturer but also has a longer term impact on patients because it puts at risk the development of new, innovative drugs, which is often cost-intensive.Citation45
To manage risks from payer coverage of new treatments while supporting early access, when possible, we first need to agree when it is reasonable to expect mature OS data and when it may not be.
Considering When Mature OS Data are, or are Not, Expected for Initial Reimbursement Decisions
There can be no simple rule as to when payers should expect mature OS data. However, considerations based on criteria such as cancer stage/disease setting, treatment efficacy and trial design can help to inform when demonstrating an OS benefit may be difficult to achieve. We have suggested considerations that might be applicable, to differing extents, across many clinical settings (). We have combined these and outlined three broad scenarios regarding expectations of mature OS data being available.
Table 2 Considerations When Proving an OS Benefit is Difficult to Achieve or is Not of Primary Relevance to Patients
Scenario 1: Median OS data are available at first pricing and reimbursement negotiations. Examples of this scenario are metastatic cancers with few treatment options, or rapidly progressing disease such as small-cell lung cancer. In such scenarios, median OS data are regarded as sufficiently mature by many payers, and final survival may be estimated through statistical extrapolation of survival curves. Other approaches to inform decisions in the absence of mature OS data are often not required in this scenario. Payers may still consider the patient relevance of non-OS endpoints, such as PFS, disease-free survival (DFS), QoL and toxicities.
Scenario 2: Limited OS data are available at first pricing and reimbursement negotiations, and mature OS data are expected within the therapy life cycle (before the therapy is off-patent or superseded). Examples based on today’s treatments are metastatic diseases with several lines of therapy still available or progressive diseases, such as locally advanced non-small-cell lung cancer (NSCLC), after chemoradiation. In such scenarios, payers should consider the patient relevance of non-OS endpoints, such as PFS, DFS, QoL and QoL related to toxicities. Other approaches include statistical inference to estimate OS, surrogacy for OS, utilizing RWE and managed entry agreements, with the choice of approach partly depending on the maturity of the data. Treatment crossover and switching can make statistical inference and other approaches more challenging.
Scenario 3: Incomplete or no OS data are available at first pricing and reimbursement decisions, or during the therapy life cycle. Examples include indolent and potentially curative cancers such as early luminal A breast cancer, and trials in which early unblinding prevents OS data maturity from being reached. In such scenarios, the intrinsic value of context-relevant and patient-relevant endpoints that reflect how the patient feels or functionsCitation47 (eg, PFS, DFS and QoL or patient-reported outcomes) will need to gain increasing weight in payer assessments. This can be as endpoints in their own right, as reflected in the ASCO Value Framework Net Health Benefit (ASCO-NHB)Citation35 and ESMO Magnitude of Clinical Benefit Scale (ESMO-MCBS),Citation36,Citation37 with assessment of which endpoints are most appropriate in a given disease context (preferably through a well-designed core outcome set).Citation48 Surrogates for OS may also be appropriate; however, approaches to assess the correlation between surrogate endpoints and OS may rely on data from similar drugs or drug classes or have high associated uncertainty.
Across these scenarios, we note the roles the product label and European Public Assessment Report (EPAR) can play in providing clear, non-technical communication of the available OS data, describing the magnitude of benefit as well as the level of immaturity or confounding. We also note the role of the manufacturer in continued OS data collection and communication after regulatory and payer approval, within or beyond the trial when feasible. We endorse the use of robust trial designs to limit bias and to achieve a high methodological quality of pivotal trials, which has been widely debated in the literature.Citation49,Citation50 This includes ensuring OS data are available for safety assessment in relevant settings.
We propose that when OS data are not available or are limited at the time of the initial pricing and reimbursement decision, this should not itself prevent patient access, provided other meaningful and patient-centered benefits have been demonstrated, which have been pre-specified and included in the trial design. We need to agree on how to appraise drugs in these scenarios, noting that the need for decision-making without mature OS data is likely to increase in the future; particularly as therapies are used in earlier disease settings or become curative. This means a specific disease setting could evolve as the science advances, from one in which mature OS data are expected at launch to one in which collection of mature OS data are not considered feasible. This development could resemble changes seen in other disease areas, for example human immunodeficiency virus (HIV)/acquired immune deficiency syndrome (AIDS) ().
Box 3 Example of a Progressive Shift Away from OS
Supporting Decision-Making in the Absence of Mature OS Data
When no or immature OS data are expected at the time of the initial reimbursement decision, alternative approaches for assessing clinical benefit are required and appropriate endpoints should be pre-specified during clinical trial design. We suggest approaches below, noting their applicability according to OS data availability. We do not expect all approaches will be used across the three scenarios described previously – for each scenario there will be a limit to how much value additional information will provide, relative to the financial impact of the decision (we note, value of information analysis can in some circumstance help to assess the expected gain from reducing uncertainty and the cost-effectiveness of further research).Citation65
Context-Relevant Endpoints
Endpoints that measure disease status and/or how patients feel and function have intrinsic value and stand “in their own right”, rather than through an association of how long the patient survives (ie, OS). For example, patients may value relief from pain, avoidance of particular symptoms or adverse events, or simply being “cancer free”, and these preferences should be ascertained for distinct patient groups. Patient-relevant endpoints can be based on tumor assessments, symptom or toxicity assessment, or QoL, with these endpoints being powered in clinical trials and/or being part of a core outcome set ().Citation48 We recommend defining the importance of context-relevant endpoints by setting (as outlined in ) through consultation with patients. We note also that statistical approaches such as generalized pairwise comparison can be used to assess net benefit across several outcomes allowing secondary endpoints a greater weight in the decision.Citation66 Furthermore, we encourage HTA agencies to define and adopt prespecified core sets of outcomes that cover a variety of target domains beyond mature OS. Asking patients what they value and quantifying the outcome provides meaningful information for decision-making and we discuss tumor assessment, QoL, symptoms and toxicity endpoints below, noting that other endpoints can be considered context relevant.
Table 3 List of Endpoints to Consider as Potential Valid Measures of Efficacy
Tumor assessment: endpoints related to disease can have a strong biological rationale – for example, showing that a tumor has responded to treatment or not progressed, and this rationale can be very specific to the setting. A demonstrable response to therapy can be associated with improved symptoms and functioning and bring other advantages, such as significant influence on quality of life in metastatic breast cancer,Citation67 or strong psychological benefit in neoadjuvant breast cancer (from achieving pathologic complete response, as reported during an HTA decision committee).Citation68,Citation69 Conversely, tumor progression was associated with substantial worsening in QoL in advanced breast, pancreatic, lung or colorectal cancer.Citation70
Similarly, event-free survival and DFS can be direct measures of clinical benefit (if toxicity does not cause detriment in QoL), for example, indicating longer time free of metastasis.Citation71
QoL, toxicity and symptoms: living better is an important potential benefit from cancer treatment, alongside the potential to live longer,Citation37 and QoL is a recognized patient-relevant endpoint in its own right.Citation31 Endpoints relating to toxicity and symptom control can also be highly relevant when measuring features of disease or treatment that are important to patients.Citation69 The analysis and interpretation of QoL measures can, however, limit comparison between trials.Citation72 For use in decision-making, QoL endpoints need to be prospectively incorporated into the trial to answer a well-defined research question, have adequate statistical power and limited missing data points, and the instrument selected according to the patient population and the objectives of treatment (eg, see considerations in ovarian cancer).Citation72–Citation75 We note that patient QoL and function can also have a profound impact on caregivers, with deteriorating functionality associated with increased caregiver burden.Citation76 When considering the toxicity of a treatment, the severity, timing and duration of adverse events can all be important, and we need to work with patients to understand these factors in different disease and treatment settings.Citation77 In some settings, patients are willing to trade significant PFS time for reductions in treatment-related toxicity, showing the importance of toxicity endpoints.Citation78
Overall, we reinforce the importance of reaching international agreement on the use of accepted patient-relevant endpoints so that prospective trials can be planned, based on criteria accepted across HTAs. The effect size for decision-making will depend on the setting and is not discussed here.
Surrogacy
Endpoints can substitute for OS or other endpoints if they are validated, ie, they have shown statistical correlation with treatment effect for the specific setting and patient population. In some settings, the validity of an endpoint as surrogate for OS has been shown;Citation81 however, surrogacy in one disease setting will rarely translate to other settings. Surrogacy is generally accepted by HTA bodies when validated for the treatment, setting and patient group of interest.Citation82 However, in practice, surrogacy for OS is often difficult to prove at the time of initial reimbursement – it is seldom possible to show a robust correlation between the treatment effect on the surrogate and the treatment effect on OS within the same treatment class and indication – especially if the new treatment is the first in class. We recommend further research into surrogate validation, focusing on sharing trial participant data and on reducing prediction uncertainty.Citation18,Citation83 Understanding which endpoints have been used, when standardized definitions are not followed, is an additional complexity. We recommend the use of standard definitions for endpoints to optimize the comparison of studies and improve surrogate analyses.
There is general agreement on validation methodology using a three-step process of establishing the level of evidence, assessing the strength of association, and quantifying the relation between the surrogate and the final outcome.Citation84 Full statistical validation is required before a surrogate for OS can be recognized, although HTA organizations’ handling of surrogate endpoints varies greatly, with inconsistency in what is considered a robust validation approach.Citation85–Citation87 Indeed, most HTA agencies do not provide guidance on how to evaluate surrogates for OS or other non-OS outcomes.Citation82 We suggest that the framework for validation requires some flexibility in settings in which clinical evidence is limited to a small number of studies, or a single study, such as can be the case for therapies that are first-in-class, for rare tumors or for specific genotypes, in which the highest level of statistical validation may not be possible.
Statistical Inference
When limited OS data are available, statistical methods can be used to adjust for uncertainty in the available evidence. Methods to adjust for treatment crossover can provide close approximations of the true treatment effect, limiting uncertainty from confounding;Citation56,Citation57 however, these techniques are by no means perfect, and will not always remove confounding variables, so they are to be used with caution. Extrapolation of the Kaplan–Meier curve for OS can estimate treatment effect beyond the time frame of the trial and is a method accepted by some HTA agencies.Citation1 There is evidence that this method can be accurate and even conservative in certain settings, with the OS benefit projections being less than the true benefit;Citation88 a study summarizing 11 cancer immunotherapy submissions to NICE found that the initial extrapolation tended to underestimate OS by 0.4–2.7%, depending on the assessment method used.Citation89 There was a similar finding when replicating an economic model that was based on extrapolated OS data (for relapsed, platinum-sensitive, BRCA mutation-positive ovarian, fallopian tube and peritoneal cancer) in which the patient benefit estimated with early data (~3.5 years of follow-up) was approximately half the patient benefit estimated when more mature data were available (~6.5 years of follow-up).Citation90
Use of Additional Evidence Gathered Outside of Clinical Trials
RWE and post-marketing data collection are becoming increasingly important when limited OS data are available from a trial (see for an example). These are expected after the initial payer decision, and at times are coupled with a managed entry agreement.Citation91,Citation92 Data collected beyond the clinical trial setting, such as through central registries, are non-randomized and can be subject to limitations, but can include more patients in the usual care setting who are potentially followed for longer than in a trial, reducing uncertainty from trial dataCitation93 and supporting treatment optimization.Citation94 Indeed, even when trial OS data are mature or not confounded, RWE remains informative for these reasons, identifying potential gaps between efficacy and effectiveness; for example, due to differences in patient characteristics and the delivery of care between settings.Citation95 An ongoing challenge is to limit delays in starting real-world studies, with the aim to start collecting RWE close to the time of regulatory approval.
Box 4 Example of Clinical Benefit Shown with RWE
RWE can also support statistical inference to build predictive models of OS according to different patient profiles, and HTA agencies are actively encouraging early dialog to align on RWE data collection.Citation96
Managed Entry Agreement Schemes
Managed entry agreements are an option to consider to grant patient access to promising therapies when uncertainty about patient benefit is high and the scheme is feasible. An example of this approach is provided by the post-2016 UK Cancer Drugs Fund, a financial program that allows initial access to a new, promising treatment while additional confirmatory evidence is being generated, often as RWE, with appraisal of this evidence a requirement for continued funding. This access is time-limited, and renewal is contingent on the manufacturer showing evidence of cost-effectiveness based on additional data. In France, the Temporary Authorization for Use (ATU) program allows access to new treatments before marketing authorization if the new therapy is for a serious or rare indication for which there are no other appropriate therapies available for in France.
Next Steps: Evolving the HTA Process to Align with Advances in Oncology and to Focus on Patient Need
We have combined the patient advocate, clinician and health economist viewpoints to suggest when and how trial outcomes other than OS could inform decision-making and how uncertainty can be addressed for investigational agents that have regulatory approval without mature OS data at the time of appraisal for reimbursement. Alignment among stakeholders is central to this and, as a first step, we suggest the following actions, with the starting point being that an absence of mature OS data is not a reason in itself to refuse patient access, providing there is robust evidence to support significant benefit to patients.
Gain agreement between payers and manufacturers on when mature OS data are to be expected for pricing and reimbursement appraisals.
Reach agreement among payers on the range of appropriate clinical endpoints (as outlined in ) that are informative in their own right – and find opportunities to engage patients in assessing the importance of these endpoints for example, in the context of core outcome sets, which should be pre-specified.
Acknowledge that the highest level of statistical validation of surrogates is unlikely to be available in all circumstances and initiate further research into trial data sharing and surrogate endpoint validation.
Manufacturers to invest in developing evidence from post-marketing RWE or statistical inference to support and extend randomized controlled trial evidence, and payers to consider this evidence.
Manufacturers and payers to use managed entry agreements when uncertainty is high and the agreement is feasible and to reappraise as more evidence becomes available.
Consult with patients to understand the value people place on benefits from new therapies, measured using a range of endpoints.
Developing operational guidance tailored by disease and treatment setting (eg, for early- versus late-stage disease) could form an additional step, requiring active collaboration between payers, manufacturers, patients and clinicians. Guidance regarding decision-making without mature OS data might also require adaptation to the specific reimbursement processes (eg, by country). This guidance could help everyone involved – payers, patients, clinicians and manufacturers – in making and understanding coverage and reimbursement decisions.
Conclusions
Our reimbursement systems should evolve to align with scientific advances in oncology. As treatments become increasingly effective and some move to being curative, we need new ways of assessing therapies without mature OS data to avoid lives being shortened unnecessarily or quality of life declining, through a lack of timely reimbursement. Fair reimbursement is important, valuing patient benefit as shown through prespecified endpoints, but reappraising as required by payers in case of ongoing uncertainty or failure to show a sustained benefit. We encourage manufacturers and payers to define flexible strategies for generating and appraising patient-relevant evidence and managing uncertainty when mature OS data are not to be expected at the time of reimbursement decision-making, and for both parties to routinely follow these strategies, as innovation is only meaningful if readily accessible to patients. We believe that the changes suggested above will promote thorough assessment and timely access to effective medicines.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The views expressed in this article are the personal views of the authors and are not made on behalf of, or reflect the position of, the companies or organizations with which the authors are affiliated. M.P.L. participated in advisory boards for AstraZeneca, Daiichi Sankyo, Gilead, Eisai, Exact Sciences, Grünenthal, Lilly, MSD, Novartis, PharmaMar, Pfizer, Pierre Fabre and Roche; performed lectures for AstraZeneca, Eisai, Exact Sciences, Lilly, MSD, Novartis, Pfizer, Gilead, Pierre Fabre, Grünenthal and Roche; and received travel expenses from Pfizer and Roche. He is an editorial board member of medactuell (medac). O.C. did not receive any funding from AstraZeneca and participated in advisory boards for BresMed. She is currently funded by the European Union’s Horizon 2020 research and innovation program under grant 779306 (COMED – Pushing the Boundaries of Cost and Outcome Analysis of Medical Technologies). W.C.N.D. is an employee of AstraZeneca. A.F. reports no competing interests. M.F. participated in and was reimbursed for advisory boards for AstraZeneca, GSK, Lilly, MSD, Novartis and Takeda; lectures for AstraZeneca; received speakers fee from GSK and ACT Genomics; received travel expenses from AstraZeneca; participated in a steering committee for AbbVie and a data monitoring committee for AGITG; and received research funding (institutional) from AstraZeneca, Beigene and Novartis. The authors report no other conflicts of interest in this work.
Acknowledgments
The authors would like to thank Bengt Liljas, Alison Horsfield, Orkideh Malkoc and Jason Ward of AstraZeneca for their critical review of the manuscript, and Polly Field and Johanna Scheinost of Oxford PharmaGenesis for medical writing support.
References
- NICE Decision Support Unit. NICE DSU technical support document 16: adjusting survival time estimates in the presence of treatment switching; 2014. Available from: http://nicedsu.org.uk/wp-content/uploads/2016/03/TSD16_Treatment_Switching.pdf. Accessed March 13, 2020.
- Prasad V. Do cancer drugs improve survival or quality of life? BMJ. 2017;359:j4528. doi:10.1136/bmj.j452828978548
- Sola-Morales O, Volmer T, Mantovani L. Perspectives to mitigate payer uncertainty in health technology assessment of novel oncology drugs. J Mark Access Health Policy. 2019;7(1):1562861. doi:10.1080/20016689.2018.156286130719243
- Pinto A, Naci H, Neez E, Mossialos E. Association between the use of surrogate measures in pivotal trials and health technology assessment decisions: a retrospective analysis of NICE and CADTH reviews of cancer drugs. Value Health. 2020;23(3):319–327. doi:10.1016/j.jval.2019.10.01032197727
- Ruof J, Knoerzer D, Dunne AA, Dintsios CM, Staab T, Schwartz FW. Analysis of endpoints used in marketing authorisations versus value assessments of oncology medicines in Germany. Health Policy (New York). 2014;118(2):242–254. doi:10.1016/j.healthpol.2014.08.004
- Chen EY-S, Joshi SK, Prasad V. FDA acceptance of surrogate endpoints in later lines of therapy. J Clin Oncol. 2018;36(15_suppl):6517. doi:10.1200/JCO.2018.36.15_suppl.6517
- Dilla T, Lizan L, Paz S, et al. Do new cancer drugs offer good value for money? The perspectives of oncologists, health care policy makers, patients, and the general population. Patient Prefer Adherence. 2015;10:1–7.26719677
- Looking to the future: oncology endpoints (summary report of a joint workshop held on of 3 July 2017 by the Academy of Medical Sciences and the Association of the British Pharmaceutical Industry); 2017. Available from: https://acmedsci.ac.uk/file-download/41135280. Accessed February 8, 2021.
- Vintura. Every day counts: improving time to patient access to innovative oncology therapies in Europe; 2020. Available from: https://www.efpia.eu/media/578013/every-day-counts.pdf. Accessed January 12, 2021.
- EFPIA. The root cause of unavailability and delay to innovative medicines: reducing the time before patients have access to innovative medicines; 2020. Available from: https://www.efpia.eu/media/554527/root-causes-unvailability-delay-cra-final-300620.pdf. Accessed April 20, 2021.
- OECD. Addressing challenges in access to oncology medicines – analytical report; 2020. Available from: https://www.oecd.org/health/health-systems/Addressing-Challenges-in-Access-to-Oncology-Medicines-Analytical-Report.pdf. Accessed April 21, 2021.
- Ciani O, Armeni P, Boscolo PR, Cavazza M, Jommi C, Tarricone R. De innovatione: the concept of innovation for medical technologies and its implications for healthcare policy-making. Health Policy Technol. 2016;5(1):47–64. doi:10.1016/j.hlpt.2015.10.005
- Hofmarcher T, Lindgren P, Wilking N, Jonsson B. The cost of cancer in Europe 2018. Eur J Cancer. 2020;129:41–49. doi:10.1016/j.ejca.2020.01.01132120274
- Food and Drug Administration. Clinical trial endpoints for the approval of cancer drugs and biologics - guidance for industry; 2018. Available from: https://www.fda.gov/media/71195/download. Accessed January 23, 2020.
- Dudley WN, Wickham R, Coombs N. An introduction to survival statistics: Kaplan-Meier analysis. J Adv Pract Oncol. 2016;7(1):91–100.27713848
- Kaufman HL, Atkins MB, Subedi P, et al. The promise of immuno-oncology: implications for defining the value of cancer treatment. J Immuno Ther Cancer. 2019;7(1):129. doi:10.1186/s40425-019-0594-0
- Dafni U. Landmark analysis at the 25-year landmark point. Circ Cardiovasc Qual Outcomes. 2011;4(3):363–371.21586725
- Ciani O, Davis S, Tappenden P, et al. Validation of surrogate endpoints in advanced solid tumors: systematic review of statistical methods, results, and implications for policy makers. Int J Technol Assess Health Care. 2014;30(3):312–324. doi:10.1017/S026646231400030025308694
- European Medicines Agency. Guideline on the evaluation of anticancer medicinal products in man; 2017. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-evaluation-anticancer-medicinal-products-man-revision-5_en.pdf. Accessed March 13, 2020.
- Kordecka A, Walkiewicz-Zarek E, Lapa J, Sadowska E, Kordecki M. Selection of endpoints in clinical trials: trends in European marketing authorization practice in oncological indications. Value Health. 2019;22(8):884–890. doi:10.1016/j.jval.2019.03.00731426929
- Enzalutamide for hormone-relapsed non-metastatic prostate cancer. NICE technology appraisal guidance [TA580]; 2019. Available from: https://www.nice.org.uk/guidance/ta580. Accessed February 1, 2021.
- Vismodegib for treating basal cell carcinoma. NICE technology appraisal guidance [TA489]; 2017. Available from: https://www.nice.org.uk/guidance/ta489. Accessed February 1, 2021.
- Justification to the resolution of the Federal Joint Committee (G-BA) on an amendment of the pharmaceuticals directive (AM-RL): Annex XII – resolutions on the benefit assessment of medicinal products with new active ingredients according to section 35a SGB V – palbociclib; 2017. Available from: https://www.g-ba.de/downloads/40-1465-4388/2017-05-18_AM-RL-XII_Palbociclib_D-264_TrG_EN.pdf. Accessed February 1, 2021.
- Justification to the resolution of the Federal Joint Committee (G-BA) on an amendment of the pharmaceuticals directive (AM-RL): Annex XII – resolutions on the benefit assessment of medicinal products with new active ingredients according to section 35a SGB V abemaciclib (breast cancer; in combination with fulvestrant); 2019. Available from: https://www.g-ba.de/downloads/40-1465-5714/2019-05-02_AM-RL-XII_Abemaciclib_D-401_TrG_EN.pdf. Accessed February 1, 2021.
- Informe de Posicionamiento Terapéutico de ixazomib (Ninlaro®) en mieloma múltiple. [Therapeutic Positioning Report of ixazomib (Ninlaro®) in multiple myeloma]; 2018. Available from: https://www.aemps.gob.es/medicamentosUsoHumano/informesPublicos/docs/IPT-ixazomib-Ninlaro-mieloma-multiple.pdf?x54046. Accessed February 1, 2021.
- Informe de Posicionamiento Terapéutico de pembrolizumab (Keytruda®) en el tratamiento de Linfoma de Hodgkin. [Report on Therapeutic Positioning of pembrolizumab (Keytruda®) in the treatment of Hodgkin's Lymphoma]; 2018. Available from: Available from: https://www.aemps.gob.es/medicamentosUsoHumano/informesPublicos/docs/IPT-pembrolizumab-Keytruda-linfoma-Hodgkin.pdf?x98091. Accessed February 1, 2021.
- Nutzenbewertungsverfahren zum Wirkstoff Bosutinib. [Benefit assessment procedure for the active ingredient bosutinib]; 2013. Available from: https://www.g-ba.de/bewertungsverfahren/nutzenbewertung/68/. Accessed January 12, 2021.
- Re-submission bosutinib; 2015. Available from: https://www.scottishmedicines.org.uk/media/1353/bosutinib__bosulif__resubmission_final_jan_2015_for_website.pdf. Accessed January 12, 2021.
- EUnetHTA. Guideline: endpoints used for relative effectiveness assessment: clinical endpoints; 2015. Available from: https://eunethta.eu/wp-content/uploads/2018/02/WP7-SG3-GL-clin_endpoints_amend2015.pdf. Accessed January 23, 2020.
- Institute for Clinical and Economic Review. ICER evidence rating matrix: a user’s guide; 2020. Available from: https://icer.org/evidence-rating-matrix/. Accessed January 6, 2020.
- Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen. General methods (version 5.0); 2017. Available from: https://www.iqwig.de/download/General-Methods_Version-5-0.pdf. Accessed January 23, 2020.
- NICE Decision Support Unit. A review of studies examining the relationship between progression-free survival and overall survival in advanced or metastatic cancer; 2012. Available from: http://nicedsu.org.uk/methods-development/pfs-os/. Accessed January 23, 2020.
- NICE Decision Support Unit. NICE DSU technical support document 20: multivariate meta-analysis of summary data for combining treatment effects on correlated outcomes and evaluating surrogate endpoints; 2019. Available from: http://nicedsu.org.uk/wp-content/uploads/2020/10/TSD-20-mvmeta-final.pdf. Accessed January 11, 2021.
- Pharmaceutical Benefits Advisory Committee Appendix 5. Translating comparative treatment effects of proposed surrogate measures to target clinical outcomes; 2016. Available from: https://pbac.pbs.gov.au/appendixes/appendix-5.html. Accessed January 20, 2020.
- Schnipper LE, Davidson NE, Wollins DS, et al. Updating the American Society of Clinical Oncology value framework: revisions and reflections in response to comments received. J Clin Oncol. 2016;34(24):2925–2934. doi:10.1200/JCO.2016.68.251827247218
- Cherny NI, Dafni U, Bogaerts J, et al. ESMO-magnitude of clinical benefit scale version 1.1. Ann Oncol. 2017;28(10):2340–2366. doi:10.1093/annonc/mdx31028945867
- Cherny NI, Sullivan R, Dafni U, et al. A standardised, generic, validated approach to stratify the magnitude of clinical benefit that can be anticipated from anti-cancer therapies: the European Society for Medical Oncology Magnitude of Clinical Benefit Scale (ESMO-MCBS). Ann Oncol. 2015;26(8):1547–1573. doi:10.1093/annonc/mdv24926026162
- NICE. Methods, processes and topic selection for health technology evaluation: proposals for change; 2021. Available from: https://www.nice.org.uk/about/what-we-do/our-programmes/nice-guidance/chte-methods-and-processes-consultation. Accessed 14 September, 2021.
- Cook JP, Golec JH, Vernon JA, Pink GH. Real option value and path dependence in oncology innovation. Int J Econ Bus. 2011;18(2):225–238. doi:10.1080/13571516.2011.584428
- Fornaro G, Federici C, Rognoni C, Ciani O. Broadening the concept of value: a scoping review on the option value of medical technologies. Value Health. 2021;24:1045–1058. doi:10.1016/j.jval.2020.12.01834243829
- FDA in brief: FDA oncologic drugs advisory committee to review status of six indications granted accelerated approval; 2021. Available from: https://www.fda.gov/news-events/fda-brief/fda-brief-fda-oncologic-drugs-advisory-committee-review-status-six-indications-granted-accelerated#:~:text=Since%20the%20inception%20of%20the,oncology%20indications%20have%20been%20withdrawn. Accessed June 15, 2021.
- Beaver JA, Pazdur R. “Dangling” accelerated approvals in oncology. N Engl J Med. 2021;384(18):e68. doi:10.1056/NEJMp210484633882220
- Rupp T, Zuckerman D. Quality of life, overall survival, and costs of cancer drugs approved based on surrogate endpoints. JAMA Intern Med. 2017;177(2):276–277. doi:10.1001/jamainternmed.2016.776127898978
- Havrilesky LJ, Lim S, Ehrisman JA, et al. Patient preferences for maintenance PARP inhibitor therapy in ovarian cancer treatment. Gynecol Oncol. 2020;156(3):561–567. doi:10.1016/j.ygyno.2020.01.02631982178
- DiMasi JA, Grabowski HG, Hansen RW. Innovation in the pharmaceutical industry: new estimates of R&D costs. J Health Econ. 2016;47:20–33. doi:10.1016/j.jhealeco.2016.01.01226928437
- Council Directive 89/105/EEC of 21 December 1988 relating to the transparency of measures regulating the prices of medicinal products for human use and their inclusion in the scope of national health insurance systems. Available from: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A31989L0105. Accessed March 16, 2021.
- Temple R. A regulatory authority’s opinion about surrogate endpoints. In: Nirnnio W, Tucker G, editors. Clinical Measurements in Drug Evaluation. John Wiley & Sons Ltd; 1995:3–22.
- Kirkham JJ, Davis K, Altman DG, et al. Core outcome Set-STAndards for development: the COS-STAD recommendations. PLoS Med. 2017;14(11):e1002447. doi:10.1371/journal.pmed.100244729145404
- Naci H, Davis C, Savović J, et al. Design characteristics, risk of bias, and reporting of randomised controlled trials supporting approvals of cancer drugs by European Medicines Agency, 2014–16: cross sectional analysis. BMJ. 2019;366:l5221. doi:10.1136/bmj.l522131533922
- Hilal T, Gonzalez-Velez M, Prasad V. Limitations in clinical trials leading to anticancer drug approvals by the US Food and Drug Administration. JAMA Intern Med. 2020;180(8):1108–1115. doi:10.1001/jamainternmed.2020.225032539071
- Fürstenau M, De Silva N, Eichhorst B, Hallek M. Minimal residual disease assessment in CLL: ready for use in clinical routine? HemaSphere. 2019;3(5):e287. doi:10.1097/HS9.000000000000028731942542
- EMA. Guideline on the use of minimal residue disease as an endpoint in chronic lymphocytic leukaemia studies; 2014. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-use-minimal-residue-disease-endpoint-chronic-lymphocytic-leukaemia-studies_en.pdf. Accessed November 11, 2020.
- Burris H, Storniolo AM. Assessing clinical benefit in the treatment of pancreas cancer: gemcitabine compared to 5-fluorouracil. Eur J Cancer. 1997;33(Suppl 1):S18–22. doi:10.1016/S0959-8049(96)00324-39166095
- Korn EL, Freidlin B, Mooney M. Stopping or reporting early for positive results in randomized clinical trials: the National Cancer Institute Cooperative Group experience from 1990 to 2005. J Clin Oncol. 2009;27(10):1712–1721. doi:10.1200/JCO.2008.19.533919237631
- AstraZeneca Press Release. Tagrisso Phase III ADAURA trial will be unblinded early after overwhelming efficacy in the adjuvant treatment of patients with EGFR-mutated lung cancer; 2020. Available from: https://www.astrazeneca.com/media-centre/press-releases/2020/tagrisso-phase-iii-adaura-trial-will-be-unblinded-early-after-overwhelming-efficacy-in-The-adjuvant-treatment-of-patients-with-egfr-mutated-lung-cancer.html. Accessed January 11, 2020.
- Ishak KJ, Proskorovsky I, Korytowsky B, Sandin R, Faivre S, Valle J. Methods for adjusting for bias due to crossover in oncology trials. PharmacoEconomics. 2014;32(6):533–546. doi:10.1007/s40273-014-0145-y24595585
- Latimer NR, Abrams KR, Lambert PC, Morden JP, Crowther MJ. Assessing methods for dealing with treatment switching in clinical trials: a follow-up simulation study. Stat Methods Med Res. 2018;27(3):765–784. doi:10.1177/096228021664226427114326
- Poveda A, Floquet A, Ledermann JA, et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): a final analysis of a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2021;22:620–631. doi:10.1016/S1470-2045(21)00073-533743851
- de Claro RA, McGinn K, Kwitkowski V, et al. U.S. Food and Drug Administration approval summary: brentuximab vedotin for the treatment of relapsed Hodgkin lymphoma or relapsed systemic anaplastic large-cell lymphoma. Clin Cancer Res. 2012;18(21):5845–5849. doi:10.1158/1078-0432.CCR-12-180322962441
- Capocaccia R, Gatta G, Dal Maso L. Life expectancy of colon, breast, and testicular cancer patients: an analysis of US-SEER population-based data. Ann Oncol. 2015;26(6):1263–1268. doi:10.1093/annonc/mdv13125735314
- Lin DY, Fischl MA, Schoenfeld DA. Evaluating the role of CD4-lymphocyte counts as surrogate endpoints in human immunodeficiency virus clinical trials. Stat Med. 1993;12(9):835–842. doi:10.1002/sim.47801209048101011
- Nakagawa F, May M, Phillips A. Life expectancy living with HIV: recent estimates and future implications. Curr Opin Infect Dis. 2013;26(1):17–25. doi:10.1097/QCO.0b013e32835ba6b123221765
- Lazarus JV, Safreed-Harmon K, Barton SE, et al. Beyond viral suppression of HIV – the new quality of life frontier. BMC Med. 2016;14(1):94. doi:10.1186/s12916-016-0640-427334606
- Hullsiek KH, Grund B. Considerations for endpoint selection when designing HIV clinical trials. Curr Infect Dis Rep. 2012;14(1):110–118. doi:10.1007/s11908-011-0231-722161272
- Wilson ECF. A practical guide to value of information analysis. PharmacoEconomics. 2015;33(2):105–121. doi:10.1007/s40273-014-0219-x25336432
- Buyse M, Saad ED, Peron J, et al. The Net Benefit of a treatment should take the correlation between benefits and harms into account. J Clin Epidemiol. 2021;137:148–158. doi:10.1016/j.jclinepi.2021.03.01833774140
- Muller V, Nabieva N, Haberle L, et al. Impact of disease progression on health-related quality of life in patients with metastatic breast cancer in the PRAEGNANT breast cancer registry. Breast. 2018;37:154–160. doi:10.1016/j.breast.2017.08.00829237546
- NICE. Pertuzumab for the neoadjuvant treatment of HER2-positive breast cancer -technology appraisal guidance [TA424]: final appraisal document; 2016. Available from: https://www.nice.org.uk/guidance/ta424/documents/final-appraisal-determination-document. Accessed March 4, 2021.
- Harbeck N, Schneeweiss A, Thuss-Patience P, et al. Neoadjuvant and adjuvant end-points in health technology assessment in oncology. Eur J Cancer. 2021;147:40–50. doi:10.1016/j.ejca.2021.01.00633611103
- Marschner N, Zacharias S, Lordick F, et al. Association of disease progression with health-related quality of life among adults with breast, lung, pancreatic, and colorectal cancer. JAMA Netw Open. 2020;3(3):e200643. doi:10.1001/jamanetworkopen.2020.064332154886
- Wu Y-L, Tsuboi M, He J, et al. Osimertinib in resected EGFR-mutated non–small-cell lung cancer. N Engl J Med. 2020;383(18):1711–1723. doi:10.1056/NEJMoa202707132955177
- Bottomley A, Pe M, Sloan J, et al. Analysing data from patient-reported outcome and quality of life endpoints for cancer clinical trials: a start in setting international standards. Lancet Oncol. 2016;17(11):e510–e514. doi:10.1016/S1470-2045(16)30510-127769798
- Wilson MK, Mercieca-Bebber R, Friedlander M. A practical guide to understanding, using and including patient reported outcomes in clinical trials in ovarian cancer. J Gynecol Oncol. 2018;29(5):e81. doi:10.3802/jgo.2018.29.e8130022641
- Joly F, Hilpert F, Okamoto A, Stuart G, Ochiai K, Friedlander M. Fifth Ovarian Cancer Consensus Conference of the Gynecologic Cancer InterGroup: recommendations on incorporating patient-reported outcomes in clinical trials in epithelial ovarian cancer. Eur J Cancer. 2017;78:133–138. doi:10.1016/j.ejca.2017.03.01928448857
- Gnanasakthy A, Barrett A, Evans E, D’Alessio D, Romano C. A review of patient-reported outcomes labeling for oncology drugs approved by the FDA and the EMA (2012–2016). Value Health. 2019;22(2):203–209. doi:10.1016/j.jval.2018.09.284230711065
- Wood R, Taylor-Stokes G, Smith F, Chaib C. The humanistic burden of advanced non-small cell lung cancer (NSCLC) in Europe: a real-world survey linking patient clinical factors to patient and caregiver burden. Qual Life Res. 2019;28(7):1849–1861. doi:10.1007/s11136-019-02152-630825160
- Friends of Cancer Research. Broadening the definition of tolerability in cancer clinical trials to better measure the patient experience; 2018. Available from: https://www.focr.org/sites/default/files/Comparative%20Tolerability%20Whitepaper_FINAL.pdf. Accessed January 12, 2021.
- Havrilesky LJ, Alvarez Secord A, Ehrisman JA, et al. Patient preferences in advanced or recurrent ovarian cancer. Cancer. 2014;120(23):3651–3659. doi:10.1002/cncr.2894025091693
- Gyawali B, Hey SP, Kesselheim AS. Evaluating the evidence behind the surrogate measures included in the FDA’s table of surrogate endpoints as supporting approval of cancer drugs. EClinicalMedicine. 2020;21:100332. doi:10.1016/j.eclinm.2020.10033232382717
- National Cancer Institute. National Cancer Institute dictionary of cancer terms. Available from: https://www.cancer.gov/publications/dictionaries/cancer-terms. Accessed May 14, 2020.
- Xie W, Regan MM, Buyse M, et al. Metastasis-free survival is a strong surrogate of overall survival in localized prostate cancer. J Clin Oncol. 2017;35(27):3097–3104. doi:10.1200/JCO.2017.73.998728796587
- Grigore B, Ciani O, Dams F, et al. Surrogate endpoints in health technology assessment: an international review of methodological guidelines. PharmacoEconomics. 2020;38(10):1055–1070. doi:10.1007/s40273-020-00935-132572825
- Powers JH, Patrick DL, Walton MK, et al. Clinician-reported outcome assessments of treatment benefit: report of the ISPOR clinical outcome assessment emerging good practices task force. Value Health. 2017;20(1):2–14. doi:10.1016/j.jval.2016.11.00528212963
- Ciani O, Buyse M, Drummond M, Rasi G, Saad ED, Taylor RS. Time to review the role of surrogate end points in health policy: state of the art and the way forward. Value Health. 2017;20(3):487–495. doi:10.1016/j.jval.2016.10.01128292495
- Buyse M, Sargent DJ, Grothey A, Matheson A, de Gramont A. Biomarkers and surrogate end points—the challenge of statistical validation. Nat Rev Clin Oncol. 2010;7(6):309–317. doi:10.1038/nrclinonc.2010.4320368727
- Prentice RL. Surrogate endpoints in clinical trials: definition and operational criteria. Stat Med. 1989;8(4):431–440. doi:10.1002/sim.47800804072727467
- Ciani O, Grigore B, Blommestein H, et al. Validity of surrogate endpoints and their impact on coverage recommendations: a retrospective analysis across International Health Technology Assessment Agencies. Med Decis Making. 2021;41:439–452.33719711
- Ouwens MJNM, Mukhopadhyay P, Zhang Y, Huang M, Latimer N, Briggs A. Estimating lifetime benefits associated with immuno-oncology therapies: challenges and approaches for overall survival extrapolations. PharmacoEconomics. 2019;37(9):1129–1138. doi:10.1007/s40273-019-00806-431102143
- Bullement A, Meng Y, Cooper M, et al. A review and validation of overall survival extrapolation in health technology assessments of cancer immunotherapy by the National Institute for Health and Care Excellence: how did the initial best estimate compare to trial data subsequently made available? J Med Econ. 2019;22(3):205–214.30422080
- Tai TA, Latimer NR, Benedict A, Kiss Z, Nikolaou A. Prevalence of immature survival data for anti-cancer drugs presented to the National Institute for Health and Care Excellence and impact on decision making. Value Health. 2021;24(4):505–512. doi:10.1016/j.jval.2020.10.01633840428
- NICE. Broader types of data to be used in development of NICE guidance; 2020. Available from: https://www.nice.org.uk/news/article/broader-types-of-data-to-be-used-in-development-of-nice-guidance. Accessed November 11, 2020.
- NICE Decision Support Unit. The use of real world data for the estimation of treatment effects in NICE decision making; 2016. Available from: http://nicedsu.org.uk/wp-content/uploads/2018/05/RWD-DSU-REPORT-Updated-DECEMBER-2016.pdf. Accessed November 11, 2020.
- Lakdawalla DN, Shafrin J, Hou N, et al. Predicting real-world effectiveness of cancer therapies using overall survival and progression-free survival from clinical trials: empirical evidence for the ASCO value framework. Value Health. 2017;20(7):866–875. doi:10.1016/j.jval.2017.04.00328712615
- Lux MP, Nabieva N, Hartkopf AD, et al. Therapy landscape in patients with metastatic HER2-positive breast cancer: data from the PRAEGNANT real-world breast cancer registry. Cancers (Basel). 2018;11(1):10. doi:10.3390/cancers11010010
- Schneeweiss A, Ettl J, Lüftner D, et al. Initial experience with CDK4/6 inhibitor-based therapies compared to antihormone monotherapies in routine clinical use in patients with hormone receptor positive, HER2 negative breast cancer - Data from the PRAEGNANT research network for the first 2 years of drug availability in Germany. Breast. 2020;54:88–95.32956934
- Annemans L, Makady A. TRUST4RD: tool for reducing uncertainties in the evidence generation for specialised treatments for rare diseases. Orphanet J Rare Dis. 2020;15(1):127. doi:10.1186/s13023-020-01370-332456653
- DeMichele A, Cristofanilli M, Brufsky A, et al. Abstract P1-19-02: overall survival for first-line palbociclib plus letrozole vs letrozole alone for HR+/HER2- metastatic breast cancer patients in US real-world clinical practice. Cancer Res. 2020;80(4Supplement):P1–19–02–P11–19–02.