Abstract
The discovery of activating mutations in the epidermal growth-factor receptor (EGFR) gene in 2004 opened a new era of personalized treatment for non-small-cell lung cancer (NSCLC). EGFR mutations are associated with a high sensitivity to EGFR tyrosine kinase inhibitors, such as gefitinib and erlotinib. Treatment with these agents in EGFR-mutant NSCLC patients results in dramatically high response rates and prolonged progression-free survival compared with conventional standard chemotherapy. Subsequently, echinoderm microtubule-associated protein-like 4 (EML4)–anaplastic lymphoma kinase (ALK), a novel driver oncogene, has been found in 2007. Crizotinib, the first clinically available ALK tyrosine kinase inhibitor, appeared more effective compared with standard chemotherapy in NSCLC patients harboring EML4-ALK. The identification of EGFR mutations and ALK rearrangement in NSCLC has further accelerated the shift to personalized treatment based on the appropriate patient selection according to detailed molecular genetic characterization. This review summarizes these genetic biomarker-based approaches to NSCLC, which allow the instigation of individualized therapy to provide the desired clinical outcome.
Introduction
Non-small-cell lung cancer (NSCLC) has a poor prognosis and remains the leading cause of death related to cancer worldwide.Citation1 For most individuals with advanced, metastatic NSCLC, cytotoxic chemotherapy is the mainstay of treatment on the basis of the associated moderate improvement in survival and quality of life.Citation2,Citation3 However, the outcome of chemotherapy in such patients has reached a plateau in terms of overall response rate (25%–35%) and overall survival (OS; 8–10 months).Citation4 This poor outcome, even for patients with advanced NSCLC who respond to such chemotherapy, has motivated a search for new therapeutic approaches.
Recent years have seen rapid progress in the development of new treatment strategies for advanced NSCLC, in particular the introduction of molecularly targeted therapies and appropriate patient selection. First, the most important change has been customization of treatment according to patient selection based on the genetic profile of the tumor. Small-molecule tyrosine kinase inhibitors (TKIs) that target the epidermal growth-factor receptor (EGFR), such as gefitinib and erlotinib, are especially effective in the treatment of NSCLC patients who harbor activating EGFR mutations. In addition, TKIs that target the receptor tyrosine kinase anaplastic lymphoma kinase (ALK) have a high response rate and markedly prolong OS in NSCLC patients positive for ALK rearrangement. The identification of EGFR mutations and ALK rearrangement in individuals with NSCLC has thus accelerated the shift to personalized treatment for this condition. Second, recent studies have demonstrated the efficacy of monoclonal antibodies, such as bevacizumab, in combination with first-line platinum-based chemotherapy in advanced non-squamous NSCLC. Third, the introduction of pemetrexed has revealed differences in OS based on histological subtype of NSCLC, with the efficacy of pemetrexed being superior to that of gemcitabine in combination with cisplatin in individuals with non-squamous NSCLC (especially adenocarcinoma), and the opposite being true for those with squamous cell carcinoma. Together, these developments show that treatment for NSCLC is evolving toward a more personalized approach based on histological subtype or the molecular or genetic profile of the tumor. This review summarizes new treatment approaches to NSCLC, focusing on the development of molecularly targeted agents, including EGFR-TKIs and ALK-TKIs, both of which are key agents for personalized (genetic information-based) therapies in individuals with this condition.
EGFR-TKIs
In 2004, three groups in the US reported the landmark findings that a subset of NSCLC patients harbor activating mutations of EGFR,Citation5–Citation7 and those tumors positive for such mutations are highly sensitive to EGFR-TKIs, such as gefitinib and erlotinib. Indeed, most NSCLC patients who experienced a marked response to EGFR-TKIs were found to harbor EGFR mutations. EGFR mutations are present predominantly in women, never-smokers, individuals with adenocarcinoma, and those of East Asian ethnicity.Citation8–Citation11 It has now been demonstrated definitively that the efficacy of EGFR-TKIs is largely dependent on the presence of an EGFR mutation in the tumor.
The role of EGFR-TKI treatment for NSCLC positive for EGFR mutations
Subsequent to the discovery of EGFR mutations in a subset of NSCLC patients in the relatively small studies published in 2004, several prospective single-arm studies showed significant efficacy of EGFR-TKIs, with a high response rate of 55%–91%, in such patients.Citation8,Citation12–Citation18 Our group analyzed individual patient data from seven prospective phase II trials of gefitinib monotherapy in Japan, including a total 148 EGFR mutation-positive patients.Citation19 The Iressa (gefitinib) Combined Analysis of Mutation Positives (I-CAMP) study showed that the overall response rate for gefitinib was 76.4%. With a median follow-up of 20.7 months, the patients treated with gefitinib showed a highly favorable progression-free survival (PFS) of 9.7 months and OS of 24.3 months. Erlotinib yielded similar results, with a median survival time of more than 2 years, in a large prospective study of the Spanish Lung Cancer Group performed with 217 EGFR mutation-positive NSCLC patients.Citation8 A pooled analysis of five additional trials also showed that EGFR mutations are a better indicator of clinical outcome in NSCLC patients than are such clinical predictors as sex, tumor histology, smoking status, and ethnicity.Citation20 These data suggested that there is no major ethnic difference in the pronounced clinical effects of EGFR-TKI treatment in EGFR mutation-positive patients, even though the EGFR mutation rate differs markedly between East Asian and Western countries, with a lower frequency in the latter.
Taken together, evidence thus supports a key role for EGFR-TKIs as a new and highly effective treatment option for NSCLC patients who harbor activating EGFR mutations. The clinical application of these findings, however, raises important issues with regard to molecular analysis of the tumor before initiation of treatment, drug selection, and treatment sequence.
The need for molecular analysis prior to treatment with EGFR-TKIs
Two pivotal phase III trials compared gefitinib with standard platinum chemotherapy in the first-line setting for individuals with advanced NSCLC.Citation21,Citation22 The patients enrolled in these trials were selected according to clinical characteristics associated with a high prevalence of activating EGFR mutations. The largest of these phase III trials, the Iressa pan-Asia Study (IPASS),Citation22 assigned 1217 East Asian never-smokers (or former light smokers) with previously untreated lung adenocarcinoma to either gefitinib or carboplatin plus paclitaxel. First-line gefitinib treatment yielded a significantly higher overall response rate and longer PFS, the primary end point of the study, compared with chemotherapy. However, the PFS curves crossed at ~6 months after the start of treatment, favoring the chemotherapy group during the initial 6 months and gefitinib thereafter, indicating that the beneficial effect of gefitinib on PFS might be limited to those patients who harbored activating EGFR mutations. A total of 683 (56%) tumor samples were obtained from the patients enrolled in this study for exploratory biomarker analysis.Citation23 EGFR mutational status was evaluated in 437 patients, of whom 261 (60%) were found to harbor an activating mutation. In comparison with chemotherapy, gefitinib treatment improved PFS in patients with EGFR mutations, whereas it was inferior to chemotherapy in those without such mutations ().Citation22 Even in a patient population selected on the basis of a clinical characteristic associated with a favorable outcome of EGFR-TKI treatment, heterogeneity was clearly apparent between the patients with or without EGFR mutations. On the other hand, there was no difference in OS between the two treatment groups for EGFR mutation-positive patients in the overall analysis ().
Table 1 Results of phase III trials comparing epidermal growth-factor receptor (EGFR)-tyrosine kinase inhibitors with chemotherapy as first-line treatment in non-small-cell lung cancer patients with EGFR mutations
The First-Line Single-Agent Iressa Versus Gemcitabine and Cisplatin Trial in Never-Smokers with Adenocarcinoma of the Lung (First-SIGNAL),Citation21 a smaller Asian trial, obtained results similar to those of the IPASS trial. The eligibility criteria for this study were also similar to those of the IPASS trial. A total of 313 Korean never-smokers with adenocarcinoma were randomized to first-line treatment with either gefitinib or gemcitabine and cisplatin. Overall, OS and PFS did not differ significantly between the two groups. Among 96 patients (31%) whose tumors were analyzed for EGFR mutations, 42 individuals (44%) were positive for such mutations (). As in the IPASS study, gefitinib prolonged PFS in EGFR mutation-positive patients, although the difference between the two treatment arms was not statistically significant. However, PFS in the EGFR mutation-negative patients was worsened by gefitinib compared with chemotherapy. In this study, there was a higher response rate to gefitinib in the mutation-negative population compared with that observed in the IPASS study, which together with the lack of a significant difference in PFS between gefitinib and chemotherapy in the mutation-positive population was due to a higher false-negative rate for EGFR mutations that resulted from non-centralized testing. The remarkable finding from both these trials, however, was that patient selection based on clinical characteristics alone was insufficient to predict accurately the benefit of EGFR-TKI treatment,Citation24 indicating that molecular analysis of EGFR mutational status is mandatory prior to treatment.
EGFR-TKIs in the first-line setting for EGFR mutation-positive patients with advanced NSCLC
The results of the IPASS study indicated that EGFR-TKIs have promising efficacy for individualized treatment of advanced NSCLC positive for EGFR mutations. We conducted a randomized phase III trial (WJTOG3405) that compared gefitinib with platinum-based chemotherapy (cisplatin plus docetaxel) in the first-line setting for 172 patients with advanced NSCLC positive for EGFR mutations.Citation25 This study met the primary end point in that PFS was found to be significantly longer in the gefitinib group than in the chemotherapy group (hazard ratio 0.489, 95% confidence interval 0.37–0.71; P < 0.0001). It did not, however, reveal a statistically significant improvement in OS in the gefitinib group. Updated OS analysis of the study showed that the median OS of patients who received gefitinib was 35.5 months, which did not differ significantly from the value of 38.8 months for those who received standard chemotherapy (hazard ratio 1.185, 95% confidence interval 0.767–1.829; P = 0.443)Citation26 (). Similar findings on the superiority of EGFR-TKIs were obtained in another Japanese phase III study (NEJ002),Citation27 in which 228 NSCLC patients with EGFR mutations were assigned to gefitinib or to carboplatin and paclitaxel. The recent updated analysis for this study again demonstrated no significant difference in OS between the two groups. Whereas the median PFS for the gefitinib group was 10.8 months compared with 5.4 months for the chemotherapy group (hazard ratio 0.322, 95% confidence interval 0.236–0.438; P < 0.001), the median OS for gefitinib and for chemotherapy was 27.7 and 26.6 months, respectively.Citation28 When survival curves are virtually identical for different groups in a randomized trial for advanced NSCLC, an improvement in quality of life or cancer-related symptoms becomes an important issue. Whereas the WJTOG3405 trial did not provide data on quality of life, the NEJ002 study found that quality of life was maintained for a longer time in patients receiving gefitinib than in those receiving standard systemic chemotherapy.Citation29 Several other phase III trials have also shown that EGFR-TKIs improve quality of life compared with chemotherapy.Citation21,Citation22,Citation30,Citation31 Superiority of erlotinib over platinum-based chemotherapy in terms of overall response rate and PFS but not OS has also been demonstrated in phase III trialsCitation30,Citation32 ().
None of the phase III trials that have compared EGFR-TKIs with standard platinum-based chemotherapy in patients with EGFR mutations has revealed a benefit in terms of OS. Failure to translate an extended PFS into an obvious survival benefit in these studies is accounted for by a high frequency of crossover treatment with EGFR-TKIs after disease progression in the chemotherapy group. However, this finding does not mean that EGFR-TKIs have little value in the first-line setting. In clinical practice, not all patients with EGFR mutations who receive standard chemotherapy in the first-line setting will be suitable for subsequent treatment with EGFR-TKIs, as a result of a rapid deterioration of their general condition and performance status due to disease progression. The chance to administer EGFR-TKIs in such patients would thus be missed. Subset analysis of the NEJ002 trial recently showed that the impact of platinum-based chemotherapy on OS was not greater than that of gefitinib in NSCLC patients with EGFR mutations.Citation28 The median OS of patients who were treated with gefitinib in any line but who did not receive platinum-based chemotherapy was more than 2 years, which is an improvement compared with historical data obtained when EGFR-TKIs were not available.Citation33 EGFR-TKIs are thus now globally recognized as important drugs and the standard first-line treatment for advanced NSCLC patients with EGFR mutations.
Resistance to EGFR-TKIs
Most NSCLC patients who harbor activating EGFR mutations, including deletions in exon 19 or the point mutation L858R in exon 21, experience an initial marked response to the EGFR-TKIs gefitinib or erlotinib. However, almost all such individuals eventually develop acquired resistance to these drugs within 1 year. In addition, 20%–30% of NSCLC patients with EGFR mutations do not show an initial response to EGFR-TKIs.Citation22,Citation25,Citation27 Therapeutic strategies to overcome EGFR-TKI resistance in NSCLC patients with EGFR mutations have been developed on the basis of the biological mechanisms of such resistance, which include a T790M secondary mutation in EGFR as well as amplification of the gene for the receptor tyrosine kinase MET, which serves as the receptor for hepatocyte growth factor.
T790M secondary EGFR mutation as a mechanism of EGFR-TKI resistance
A secondary point mutation of EGFR that results in the substitution of methionine for threonine at amino acid position 790 (T790M) was the first identified mechanism of acquired EGFR-TKI resistance in NSCLC patients.Citation34,Citation35 About 50%–70% of NSCLC patients who develop acquired resistance to EGFR-TKIs have been found to harbor the T790M secondary mutation, with the mutation not being present in tumor specimens obtained before EGFR-TKI treatment.Citation36,Citation37 The T790M mutation has also been detected, however, in a small proportion of NSCLC patients who have not yet received any treatment.Citation38 Several highly sensitive methods, compared with direct sequencing, have recently been developed to detect a low frequency of T790M in genetically heterogeneous clinical specimens. These methods include polymerase chain reaction (PCR) invader,Citation39 peptide nucleic acid-locked nucleic acid PCR clamp,Citation40 and Cycleave PCR assays.Citation37 The Scorpion amplification-refractory mutation system assay identified T790M in circulating tumor cells of NSCLC patients before EGFR-TKI treatment.Citation41 Matrix-assisted laser desorption ionization–time of flight mass spectrometry detected T790M in 25.2% of TKI-naive NSCLC patients who harbored activating EGFR mutations.Citation42 The presence of the T790M mutation in NSCLC patients before treatment was found to be associated with a significantly shorter PFS after initiation of EGFR-TKI treatment.Citation41,Citation42 These observations thus suggest that T790M contributes not only to acquired resistance to EGFR-TKIs but also to intrinsic resistance to these drugs.
Similar to mutations in BCR-ABL (T315I)Citation43 or in KIT (T670I)Citation44 that underlie resistance to imatinib, T790M is thought to interfere with the binding of EGFR-TKIs at the adenosine triphosphate-binding cleft of EGFR.Citation34,Citation35,Citation45 On the other hand, the affinity of this cleft for adenosine triphosphate is increased by T790M.Citation46 Treatment with irreversible EGFR-TKIs such as afatinib (BIBW2992)Citation47 and dacomitinib (PF00299804)Citation48,Citation49 is thought to be a potential approach to overcome the resistance conferred by T790M (). Although recent preclinical studies have demonstrated only limited activity of irreversible EGFR-TKIs alone in NSCLC cells positive for T790M,Citation50,Citation51 such studies have shown that combinations of these drugs with other agents – such as afatinib combined with cetuximab (a monoclonal antibody to EGFR)Citation52 or with PI-103 (an inhibitor of signaling by phosphoinositide 3-kinase and mammalian target of rapamycin)Citation50 – are more promising as a treatment option to overcome resistance conferred by T790M. In addition, treatment with heat-shock protein 90 inhibitors such as 17-DMAG is also thought to be a potential approach to counter the effect of T790M.Citation53 Furthermore, WZ4002, which selectively inhibits the activity of EGFR harboring activating mutations and T790M, has been identified as a candidate for translation to the clinic.Citation54
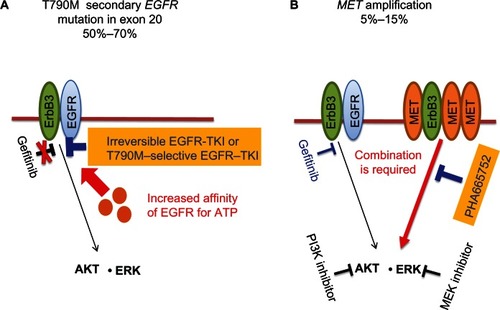
Figure 1 (A and B) Strategies to overcome acquired epidermal growth-factor receptor (EGFR)-tyrosine kinase inhibitor (TKI) resistance in non-small-cell lung cancer (NSCLC). (A) The T790M secondary mutation in exon 20 of EGFR is present in 50%–70% of NSCLC patients who acquire resistance to EGFR-TKIs, such as gefitinib or erlotinib. In such patients, gefitinib is not able to compete with adenosine triphosphate (ATP) for binding to the ATP-binding cleft of EGFR because of an increased affinity of this site for ATP. Treatment with irreversible EGFR-TKIs or EGFR-TKIs selective for EGFR harboring T790M is thus thought to represent a potential approach to overcome the resistance conferred by this mutation. (B) Amplification of MET is apparent in 5%–15% of NSCLC patients who acquire EGFR-TKI resistance. In this situation, MET signaling through ErbB3 (HER3) is activated in addition to EGFR signaling, with the result that both gefitinib and a MET inhibitor (such as PHA665752) are necessary to overcome the resistance conferred by MET amplification. The combination of inhibitors that block molecules that function downstream of both EGFR and MET, such as a phosphoinositide 3-kinase (PI3K) inhibitor combined with an MEK (ERK kinase) inhibitor, might also be an alternative approach to overcome the resistance induced by MET amplification.
MET amplification and other mechanisms of EGFR-TKI resistance
MET amplification was identified as a mechanism of gefitinib resistance in 22% of NSCLC patients with acquired resistance to this drug.Citation55 Both MET and EGFR signaling were found to activate phosphoinositide 3-kinase via ErbB3 (also known as HER3) in gefitinib-resistant NSCLC cells positive for MET amplification.Citation55 The combination of gefitinib and the MET inhibitor PHA665752 was thus required to block survival signaling in these cells ().Citation55 In the clinical setting, a phase I/II trial of the MET-TKI crizotinib in combination with erlotinib is ongoing in patients with NSCLC (NCT00965731). The results of the phase I portion of a phase I/II dose-finding study for crizotinib were reported at the 2012 Annual Meeting of the European Society for Medical Oncology.Citation56 This study allowed recruitment of NSCLC patients who had received prior EGFR-TKI therapy. Of the 25 enrolled patients, two showed a partial response and eight had stable disease. Although few studies have addressed other therapies to overcome resistance conferred by MET amplification, dasatinib, an inhibitor of the non-receptor tyrosine kinase Src, effectively inhibited the growth of gefitinib-resistant NSCLC cells positive for EGFR mutation and MET amplification, with this approach being based on the observation that Src acts downstream of both EGFR and MET in these cells.Citation57 Another preclinical study suggested that combination of gefitinib with the oral fluoropyrimidine derivative S-1 is a potential therapy to overcome acquired resistance due to MET amplification.Citation58
Several other possible mechanisms of acquired resistance to EGFR-TKIs – including upregulation of insulin-like growth-factor 1 receptor signaling,Citation59–Citation61 loss of the phosphatase PTEN,Citation62,Citation63 small-cell transformation,Citation64–Citation66 and overexpression of hepatocyte growth factorCitation67 – have been described. However, the mechanism of acquired EGFR-TKI resistance in ~30% of NSCLC patients remains unclear. Recent studies have implicated epithelial–mesenchymal transition (EMT) as a possible mechanism of acquired EGFR-TKI resistance in NSCLC cell lines.Citation68,Citation69 Furthermore, tumor cells having undergone EMT were detected in a subset of NSCLC patients who developed EGFR-TKI resistance.Citation70,Citation71 Although histone deacetylase inhibitors may help overcome EMT-related resistance to EGFR-TKIs in NSCLC,Citation69 further studies are required to provide a better understanding of the role of EMT in such resistance and to identify novel therapeutic strategies to overcome it.
ALK inhibitors
Role of the EML4-ALK fusion gene in NSCLC
The ALK gene undergoes transforming rearrangements that generate fusion genes in several human hematologic and solid malignancies.Citation72,Citation73 The partner genes in such fusions with ALK include NPM, TPM3, and CLTC. In 2007, Soda et alCitation74 identified a new ALK rearrangement that results from an inversion within chromosome 2p and generates a transforming fusion of ALK with the echinoderm microtubule-associated protein-like 4 gene (EML4) in NSCLC. Several in-frame fusion variants of EML4-ALK have been found to be generated as a result of diversity in the breakpoint–fusion point of EML4, and other rare non-EML4 fusion partners of ALK, including KIF5B and TFG, have since been identified in NSCLC.Citation75–Citation77
The reported incidence of EML4-ALK has varied among studies as a result of different procedures adopted for detection of the fusion gene.Citation78 The prevalence of ALK rearrangement as detected either by reverse transcription and PCR analysis or by fluorescence in situ hybridization (FISH) is relatively low (in the order of 3%–5%), however, in unselected patients with NSCLC.Citation78 ALK status as determined by FISH, which is regarded as the global standard for detection of ALK rearrangement, is currently considered the key predictive marker for treatment of NSCLC patients with an ALK-TKI.Citation79 Indeed, positive identification of EML4-ALK by FISH was associated with a high sensitivity to a small-molecule inhibitor of ALK tyrosine kinase activity in a clinical trial.Citation79 Although EML4-ALK is present in only a small proportion of unselected NSCLC patients, the clinical characteristics of NSCLC patients harboring EML4-ALK are highly similar to those of such patients who harbor activating EGFR mutations.Citation76,Citation79,Citation80 Both types of gene alteration are thus found most frequently in patients with adenocarcinoma and in those who are never- or light smokers. Individuals with EML4-ALK also tend to be younger than unselected NSCLC patients. With rare exceptions, the presence of EML4-ALK appears to be mutually exclusive with that of EGFR or KRAS mutations.Citation81 Furthermore, whereas EGFR mutations are present more frequently in East Asian populations than in Caucasians, no ethnic differences in the frequency of EML4-ALK among NSCLC patients have been reported. With regard to its associated pathological features, EML4-ALK tends to be found in lung adenocarcinoma with a mucinous cribriform pattern and signet-ring cells,Citation82–Citation84 although it has also been detected in other pathological subtypes of NSCLC and other types of cancer, including breast and colorectal tumors.Citation85,Citation86
Crizotinib
Whereas several ALK inhibitors have already been introduced into clinical trials, crizotinib was the first ALK-TKI to be so evaluated. Crizotinib was initially designed as an inhibitor of MET and is thus also known as a dual inhibitor of both ALK and MET kinases.Citation87,Citation88 A phase I trial revealed marked therapeutic efficacy of crizotinib in patients with NSCLC positive for EML4-ALK (), with an overall response rate of 60.8% and a disease control rate of up to 90%.Citation79,Citation89 Crizotinib was thus approved as a therapeutic drug for certain patients with NSCLC positive for EML4-ALK by the US Food and Drug Administration.
Table 2 Clinical trials of crizotinib treatment for advanced non-small-cell lung cancer positive for anaplastic lymphoma kinase rearrangement
These remarkable findings of crizotinib activity led to two subsequent phase III clinical trials of this drug. The Profile 1007 trial, a global, randomized phase III study with a primary end point of PFS, was designed to compare crizotinib with pemetrexed or docetaxel chemotherapy in patients with advanced NSCLC positive for EML4-ALK in the second-line setting.Citation90 Patients treated with docetaxel or pemetrexed who developed progressive disease were allowed to receive crizotinib in a companion phase II trial (Profile 1005) that was designed to enroll EML4-ALK-positive NSCLC patients who were not eligible for Profile 1007.Citation91 The purpose of the phase II study was to evaluate crizotinib in terms of efficacy and safety through analysis of adverse events. The second phase III trial, Profile 1014, is ongoing and compares crizotinib with cisplatin/carboplatin and pemetrexed as a first-line treatment for patients with advanced NSCLC harboring EML4-ALK as detected by FISH (NCT01154140). The primary end point of this trial is also PFS, as was the case for EGFR-TKI trials, because of a high potential for crossover treatment. The Profile 1007 trial showed that crizotinib significantly prolonged PFS and had a higher overall response rate in comparison with standard single-agent chemotherapy (). However, no statistically significant difference in OS was observed between crizotinib and chemotherapy as a result of the anticipated high level of crossover, although the interim analysis of OS was premature. Furthermore, the improvement in both lung cancer-related symptoms and quality of life relative to baseline observed with crizotinib was significantly greater than that achieved with chemotherapy. These findings thus demonstrated an efficacy for crizotinib in patients with ALK rearrangement-positive NSCLC similar to that for EGFR-TKIs in those with EGFR mutation-positive NSCLC. These thus established crizotinib treatment as the standard of care for previously treated patients with advanced NSCLC harboring EML4-ALK.
Other ALK inhibitors
As of March 2013, at least five additional distinct inhibitors of ALK were undergoing evaluation in early clinical trials worldwide (). CH5424802 is a potent, selective inhibitor of ALK that was found to show preferential antitumor activity for ALK rearrangement-positive cancer cells, such as EML4-ALK-positive NSCLC cells and NPM-ALK-positive anaplastic large-cell lymphoma cells, in a preclinical study.Citation92 This agent also potently inhibits the activity of ALK containing the L1196M “gatekeeper mutation,” which confers clinical resistance to ALK inhibitors. A phase I trial revealed that CH5424802 had marked efficacy and was well tolerated in patients with ALK rearrangement-positive NSCLC.Citation93 Preliminary results of a phase II trial have also been presented.Citation94 As of March 2012, 34 patients with ALK rearrangement-positive NSCLC previously untreated with ALK inhibitors had been enrolled and received CH5424802 at 300 mg twice daily until the development of progressive disease or unacceptable toxicity. Most of the patients were never-smokers with good performance status and had previously received extensive chemotherapy. Of the first 15 patients receiving this agent, one individual achieved a complete response and ten individuals showed a partial response, yielding a response rate of 73.3%. Most treatment-related adverse events were of grade 1. No dose reductions were necessary, although two cases of grade 3 neutropenia occurred. With regard to eye disorders, which are frequently observed with crizotinib, only one case of grade 1 was described.
Table 3 Anaplastic lymphoma kinase (ALK) inhibitors in early clinical development for treatment of ALK rearrangement-positive non-small-cell lung cancer
LDK378 is another potent and selective small-molecule ALK inhibitor (median inhibitory concentration of 0.00015 μM) that does not inhibit MET. A phase I trial has been initiated to determine the maximum tolerated dose and safety profile of this agent in patients with solid tumors positive for genetic alterations of ALK, including gene rearrangement.Citation95 Patients previously untreated with ALK inhibitors or those with disease relapse after previous treatment with such an inhibitor are eligible for enrollment in the trial. As of August 2012, 79 ALK rearrangement-positive NSCLC patients, including 56 who had received prior crizotinib treatment, were enrolled and had been administered LDK378 in a dose range of 50–750 mg/day. At doses of ≥400 mg, steady-state concentrations of the drug in plasma exceeded efficacious levels determined in vivo. Among 59 evaluable NSCLC patients, there were 24 responses, yielding a response rate of 40.7%. Among 45 patients with ALK rearrangement-positive NSCLC who had experienced progression following crizotinib treatment and received LDK378 at ≥400 mg/day, preliminary responses were apparent in 21 patients (46.7%). Dose-limiting toxicity occurred at doses of ≥400 mg/day and included diarrhea, nausea, vomiting, dehydration, and elevation of alanine aminotransferase levels. The maximum tolerated dose was 750 mg/day. These preliminary results suggest that LDK378 may prove effective for the treatment of patients with ALK rearrangement-positive NSCLC who develop acquired resistance to crizotinib. Two phase II trials of LDK378 at a dose of 750 mg/day are ongoing for ALK rearrangement-positive NSCLC patients previously treated either with crizotinib or with standard chemotherapy.
AP26113 is a synthetic and highly selective small-molecule inhibitor designed to target ALK. It is actually a dual inhibitor for both ALK and EGFR with activating mutations, whereas it does not inhibit wild-type EGFR. Furthermore, it potently inhibits TKI-resistant forms of these kinases harboring gatekeeper mutations (L1196M in ALK and T790M in EGFR).Citation96,Citation97 As of September 2012, 15 patients with advanced malignancies, including eleven individuals with NSCLC, had been enrolled in a phase I/II trial of AP26113.Citation98 The NSCLC patients included four with ALK rearrangement-positive tumors that developed resistance to prior crizotinib treatment. All patients received AP26113 at doses of 30–120 mg/day. All four ALK rearrangement-positive NSCLC patients showed partial responses, with one patient treated at a dose of 60 mg and the other three at 90 mg. The efficacy and safety of this agent were evaluated at a dose of 120 mg. Neither dose-limiting toxicities nor treatment-related serious adverse events were observed, with the most common adverse events being fatigue and nausea. The results of the phase I portion of the trial thus indicate that AP26113 is effective with acceptable toxicity, and they suggest it may have potential for the treatment of patients with ALK rearrangement-positive NSCLC who have experienced disease progression while receiving crizotinib. The phase II portion will evaluate the efficacy and safety of AP26113 in several cohorts.
Phase I clinical trials of the remaining two ALK-specific agents – X-396Citation99 and ASP3026Citation100 – have been initiated in patients with ALK rearrangement-positive advanced solid tumors and NSCLC, respectively.
Conclusion
Over the course of the last several years, the introduction of molecularly targeted agents such as gefitinib, erlotinib, and crizotinib has resulted in marked changes in treatment approaches to NSCLC. The introduction of these agents into the clinic has followed the identification of genetic changes that give rise to NSCLC and has been accompanied by appropriate patient selection. Such drugs are expensive, however, and their use is limited to subsets of patients in whom the target has undergone activating changes and become an “essential growth driver” for the cancer. Personalized therapy is based on the notion that appropriate patient selection according to detailed molecular genetic characterization will allow the instigation of individualized therapy to provide the desired clinical outcome. Treatment of NSCLC will thus come to rely more and more on a genetic biomarker-based approach to prolong survival. The advent of next-generation technologies for genetic characterization of each patient will be important to facilitate further development of individualized treatment of NSCLC with molecularly targeted agents.
Disclosure
The authors report no conflicts of interest in this work.
References
- SiegelRNaishadhamDJemalACancer statistics, 2012CA Cancer J Clin201262102922237781
- AzzoliCGBakerSJrTeminS[American Society of Clinical Oncology Clinical Practice Guideline update on chemotherapy for stage IV non-small-cell lung cancer.]Zhongguo Fei Ai Za Zhi200913171189 Chinese20681066
- D’AddarioGFruhMReckMBaumannPKlepetkoWFelipEMetastatic non-small-cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-upAnn Oncol201021Suppl 5v116v11920555059
- National Comprehensive Cancer NetworkNCCN Clinical Practice Guidelines in Oncology: non-small cell lung cancer. Version 2.2013 Available from: http://www.nccn.org/professionals/physician_gls/pdf/nscl.pdfAccessed March 21, 2013
- LynchTJBellDWSordellaRActivating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinibN Engl J Med20043502129213915118073
- PaezJGJannePALeeJCEGFR mutations in lung cancer: correlation with clinical response to gefitinib therapyScience20043041497150015118125
- PaoWMillerVZakowskiMEGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinibProc Natl Acad Sci USA2004101133061331115329413
- RosellRMoranTQueraltCScreening for epidermal growth factor receptor mutations in lung cancerN Engl J Med200936195896719692684
- Cortes-FunesHGomezCRosellREpidermal growth factor receptor activating mutations in Spanish gefitinib-treated non-small-cell lung cancer patientsAnn Oncol2005161081108615851406
- MitsudomiTKosakaTEndohHMutations of the epidermal growth factor receptor gene predict prolonged survival after gefitinib treatment in patients with non-small-cell lung cancer with postoperative recurrenceJ Clin Oncol2005232513252015738541
- HanSWKimTYJeonYKOptimization of patient selection for gefitinib in non-small cell lung cancer by combined analysis of epidermal growth factor receptor mutation, K-ras mutation, and Akt phosphorylationClin Cancer Res2006122538254416638863
- InoueASuzukiTFukuharaTProspective phase II study of gefitinib for chemotherapy-naive patients with advanced non-small-cell lung cancer with epidermal growth factor receptor gene mutationsJ Clin Oncol2006243340334616785471
- SugioKUramotoHOnitsukaTProspective phase II study of gefitinib in non-small cell lung cancer with epidermal growth factor receptor gene mutationsLung Cancer20096431431818992959
- SunagaNTomizawaYYanagitaniNPhase II prospective study of the efficacy of gefitinib for the treatment of stage III/IV non-small cell lung cancer with EGFR mutations, irrespective of previous chemotherapyLung Cancer20075638338917368623
- SutaniANagaiYUdagawaKGefitinib for non-small-cell lung cancer patients with epidermal growth factor receptor gene mutations screened by peptide nucleic acid-locked nucleic acid PCR clampBr J Cancer2006951483148917106442
- TamuraKOkamotoIKashiiTMulticentre prospective phase II trial of gefitinib for advanced non-small cell lung cancer with epidermal growth factor receptor mutations: results of the West Japan Thoracic Oncology Group trial (WJTOG0403)Br J Cancer20089890791418283321
- YoshidaKYatabeYParkJYProspective validation for prediction of gefitinib sensitivity by epidermal growth factor receptor gene mutation in patients with non-small cell lung cancerJ Thorac Oncol20072222817410005
- SequistLVMartinsRGSpigelDFirst-line gefitinib in patients with advanced non-small-cell lung cancer harboring somatic EGFR mutationsJ Clin Oncol2008262442244918458038
- MoritaSOkamotoIKobayashiKCombined survival analysis of prospective clinical trials of gefitinib for non-small cell lung cancer with EGFR mutationsClin Cancer Res2009154493449819531624
- JackmanDMMillerVACioffrediLAImpact of epidermal growth factor receptor and KRAS mutations on clinical outcomes in previously untreated non-small cell lung cancer patients: results of an online tumor registry of clinical trialsClin Cancer Res2009155267527319671843
- HanJYParkKKimSWFirst-SIGNAL: first-line single-agent Iressa versus gemcitabine and cisplatin trial in never-smokers with adenocarcinoma of the lungJ Clin Oncol2012301122112822370314
- MokTSWuYLThongprasertSGefitinib or carboplatin-paclitaxel in pulmonary adenocarcinomaN Engl J Med200936194795719692680
- FukuokaMWuYLThongprasertSBiomarker analyses and final overall survival results from a phase III, randomized, open-label, first-line study of gefitinib versus carboplatin/paclitaxel in clinically selected patients with advanced non-small-cell lung cancer in Asia (IPASS)J Clin Oncol2011292866287421670455
- GridelliCDe MarinisFDi MaioMCortinovisDCappuzzoFMokTGefitinib as first-line treatment for patients with advanced non-small-cell lung cancer with activating epidermal growth factor receptor mutation: implications for clinical practice and open issuesLung Cancer2011723821216488
- MitsudomiTMoritaSYatabeYGefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trialLancet Oncol20101112112820022809
- MitsudomiTMoritaSYatabeYUpdated overall survival results of WJTOG 3405, a randomized phase III trial comparing gefitinib (G) with cisplatin plus docetaxel (CD) as the first-line treatment for patients with non-small cell lung cancer harboring mutations of the epidermal growth factor receptor (EGFR)J Clin Oncol201230Suppl7521
- MaemondoMInoueAKobayashiKGefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFRN Engl J Med20103622380238820573926
- InoueAKobayashiKMaemondoMUpdated overall survival results from a randomized phase III trial comparing gefitinib with carboplatin-paclitaxel for chemo-naive non-small cell lung cancer with sensitive EGFR gene mutations (NEJ002)Ann Oncol201324545922967997
- OizumiSKobayashiKInoueAQuality of life with gefitinib in patients with EGFR-mutated non-small cell lung cancer: quality of life analysis of North East Japan Study Group 002 TrialOncologist20121786387022581822
- ZhouCWuYLChenGErlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutationpositive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 studyLancet Oncol20111273574221783417
- YangJCHSchulerMHYamamotoNLUX-Lung 3: a randomized, open-label, phase III study of afatinib versus pemetrexed and cisplatin as first-line treatment for patients with advanced adenocarcinoma of the lung harboring EGFR-activating mutationsJ Clin Oncol201230SupplLBA7500
- RosellRCarcerenyEGervaisRErlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trialLancet Oncol20121323924622285168
- TakanoTFukuiTOheYEGFR mutations predict survival benefit from gefitinib in patients with advanced lung adenocarcinoma: a historical comparison of patients treated before and after gefitinib approval in JapanJ Clin Oncol2008265589559518794545
- KobayashiSBoggonTJDayaramTEGFR mutation and resistance of non-small-cell lung cancer to gefitinibN Engl J Med200535278679215728811
- PaoWMillerVAPolitiKAAcquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domainPLoS Med20052e7315737014
- BalakMNGongYRielyGJNovel D761Y and common secondary T790M mutations in epidermal growth factor receptor-mutant lung adenocarcinomas with acquired resistance to kinase inhibitorsClin Cancer Res2006126494650117085664
- KosakaTYatabeYEndohHAnalysis of epidermal growth factor receptor gene mutation in patients with non-small cell lung cancer and acquired resistance to gefitinibClin Cancer Res2006125764576917020982
- ToyookaSKiuraKMitsudomiTEGFR mutation and response of lung cancer to gefitinibN Engl J Med20053522136 author reply 213615901872
- HallJGEisPSLawSMSensitive detection of DNA polymorphisms by the serial invasive signal amplification reactionProc Natl Acad Sci USA2000978272827710890904
- NagaiYMiyazawaHHuqunGenetic heterogeneity of the epidermal growth factor receptor in non-small cell lung cancer cell lines revealed by a rapid and sensitive detection system, the peptide nucleic acid-locked nucleic acid PCR clampCancer Res2005657276728216105816
- MaheswaranSSequistLVNagrathSDetection of mutations in EGFR in circulating lung-cancer cellsN Engl J Med200835936637718596266
- SuKYChenHYLiKCPretreatment epidermal growth factor receptor (EGFR) T790M mutation predicts shorter EGFR tyrosine kinase inhibitor response duration in patients with non-small-cell lung cancerJ Clin Oncol20123043344022215752
- BranfordSRudzkiZWalshSHigh frequency of point mutations clustered within the adenosine triphosphate-binding region of BCR/ABL in patients with chronic myeloid leukemia or Ph-positive acute lymphoblastic leukemia who develop imatinib (STI571) resistanceBlood2002993472347511964322
- TamboriniEBonadimanLGrecoAA new mutation in the KIT ATP pocket causes acquired resistance to imatinib in a gastrointestinal stromal tumor patientGastroenterology200412729429915236194
- KumarAPetriETHalmosBBoggonTJStructure and clinical relevance of the epidermal growth factor receptor in human cancerJ Clin Oncol2008261742175118375904
- YunCHMengwasserKETomsAVThe T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATPProc Natl Acad Sci USA20081052070207518227510
- LiDAmbrogioLShimamuraTBIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer modelsOncogene2008274702471118408761
- EngelmanJAZejnullahuKGaleCMPF00299804, an irreversible pan-ERBB inhibitor, is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinibCancer Res200767119241193218089823
- GonzalesAJHookKEAlthausIWAntitumor activity and pharmacokinetic properties of PF-00299804, a second-generation irreversible pan-erbB receptor tyrosine kinase inhibitorMol Cancer Ther200871880188918606718
- SosMLRodeHBHeynckSChemogenomic profiling provides insights into the limited activity of irreversible EGFR inhibitors in tumor cells expressing the T790M EGFR resistance mutationCancer Res20107086887420103621
- TakezawaKOkamotoITanizakiJEnhanced anticancer effect of the combination of BIBW2992 and thymidylate synthase-targeted agents in non-small cell lung cancer with the T790M mutation of epidermal growth factor receptorMol Cancer Ther201091647165620530710
- RegalesLGongYShenRDual targeting of EGFR can overcome a major drug resistance mutation in mouse models of EGFR mutant lung cancerJ Clin Invest20091193000301019759520
- ShimamuraTLiDJiHHsp90 inhibition suppresses mutant EGFR-T790M signaling and overcomes kinase inhibitor resistanceCancer Res2008685827583818632637
- ZhouWErcanDChenLNovel mutant-selective EGFR kinase inhibitors against EGFR T790MNature20094621070107420033049
- EngelmanJAZejnullahuKMitsudomiTMET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signalingScience20073161039104317463250
- OuSHIGovindanREatonKDPhase I/II dose-finding study of crizotinib (criz) in combination with erlotinib (E) in patients (pts) with advanced non-small cell lung cancer (NSCLC)J Clin Oncol201230Suppl2610
- YoshidaTOkamotoIOkamotoWEffects of Src inhibitors on cell growth and epidermal growth factor receptor and MET signaling in gefitinib-resistant non-small cell lung cancer cells with acquired MET amplificationCancer Sci201010116717219804422
- OkabeTOkamotoITsukiokaSAddition of S-1 to the epidermal growth factor receptor inhibitor gefitinib overcomes gefitinib resistance in non-small cell lung cancer cell lines with MET amplificationClin Cancer Res20091590791319188161
- MorgilloFWooJKKimESHongWKLeeHYHeterodimerization of insulin-like growth factor receptor/epidermal growth factor receptor and induction of survivin expression counteract the antitumor action of erlotinibCancer Res200666101001011117047074
- MorgilloFKimWYKimESCiardielloFHongWKLeeHYImplication of the insulin-like growth factor-IR pathway in the resistance of non-small cell lung cancer cells to treatment with gefitinibClin Cancer Res2007132795280317473213
- GuixMFaberACWangSEAcquired resistance to EGFR tyrosine kinase inhibitors in cancer cells is mediated by loss of IGF-binding proteinsJ Clin Invest20081182609261918568074
- SosMLKokerMWeirBAPTEN loss contributes to erlotinib resistance in EGFR-mutant lung cancer by activation of Akt and EGFRCancer Res2009693256326119351834
- YamasakiFJohansenMJZhangDAcquired resistance to erlotinib in A-431 epidermoid cancer cells requires down-regulation of MMAC1/PTEN and up-regulation of phosphorylated AktCancer Res2007675779578817575145
- ZakowskiMFLadanyiMKrisMGMemorial Sloan-Kettering Cancer Center Lung Cancer OncoGenome GroupEGFR mutations in small-cell lung cancers in patients who have never smokedN Engl J Med200635521321516837691
- ArcilaMEOxnardGRNafaKRebiopsy of lung cancer patients with acquired resistance to EGFR inhibitors and enhanced detection of the T790M mutation using a locked nucleic acid-based assayClin Cancer Res2011171169118021248300
- MorinagaROkamotoIFurutaKSequential occurrence of non-small cell and small cell lung cancer with the same EGFR mutationLung Cancer20075841141317601631
- YanoSWangWLiQHepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor-activating mutationsCancer Res2008689479948719010923
- YaoZFenoglioSGaoDCTGF-beta IL-6 axis mediates selective and adaptive mechanisms of resistance to molecular targeted therapy in lung cancerProc Natl Acad Sci USA2010107155351554020713723
- SudaKTomizawaKFujiiMEpithelial to mesenchymal transition in an epidermal growth factor receptor-mutant lung cancer cell line with acquired resistance to erlotinibJ Thorac Oncol201161152116121597390
- UramotoHShimokawaHHanagiriTKuwanoMOnoMExpression of selected gene for acquired drug resistance to EGFR-TKI in lung adenocarcinomaLung Cancer20113025132517
- SequistLVWaltmanBADias-SantagataDGenotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitorsSci Transl Med2011375ra26
- PalmerRHVernerssonEGrabbeCHallbergBAnaplastic lymphoma kinase: signalling in development and diseaseBiochem J200942034536119459784
- ManoHNon-solid oncogenes in solid tumors: EML4-ALK fusion genes in lung cancerCancer Sci2008992349235519032370
- SodaMChoiYLEnomotoMIdentification of the transforming EML4-ALK fusion gene in non-small-cell lung cancerNature200744856156617625570
- ChoiYLTakeuchiKSodaMIdentification of novel isoforms of the EML4-ALK transforming gene in non-small cell lung cancerCancer Res2008684971497618593892
- TakeuchiKChoiYLSodaMMultiplex reverse transcription-PCR screening for EML4-ALK fusion transcriptsClin Cancer Res2008146618662418927303
- TakeuchiKChoiYLTogashiYKIF5B-ALK, a novel fusion oncokinase identified by an immunohistochemistry-based diagnostic system for ALK-positive lung cancerClin Cancer Res2009153143314919383809
- SolomonBVarella-GarciaMCamidgeDRALK gene rearrangements: a new therapeutic target in a molecularly defined subset of non-small cell lung cancerJ Thorac Oncol200941450145420009909
- KwakELBangYJCamidgeDRAnaplastic lymphoma kinase inhibition in non-small-cell lung cancerN Engl J Med20103631693170320979469
- WongDWLeungELSoKKThe EML4-ALK fusion gene is involved in various histologic types of lung cancers from nonsmokers with wild-type EGFR and KRASCancer20091151723173319170230
- TiseoMGelsominoFBoggianiDEGFR and EML4-ALK gene mutations in NSCLC: a case report of erlotinib-resistant patient with both concomitant mutationsLung Cancer20117124124321168933
- Mino-KenudsonMChirieacLRLawKA novel, highly sensitive antibody allows for the routine detection of ALK-rearranged lung adenocarcinomas by standard immunohistochemistryClin Cancer Res2010161561157120179225
- RodigSJMino-KenudsonMDacicSUnique clinicopathologic features characterize ALK-rearranged lung adenocarcinoma in the western populationClin Cancer Res2009155216522319671850
- YoshidaATsutaKNakamuraHComprehensive histologic analysis of ALK-rearranged lung carcinomasAm J Surg Pathol2011351226123421753699
- LinELiLGuanYExon array profiling detects EML4-ALK fusion in breast, colorectal, and non-small cell lung cancersMol Cancer Res200971466147619737969
- SugawaraETogashiYKurodaNIdentification of anaplastic lymphoma kinase fusions in renal cancer: large-scale immunohistochemical screening by the intercalated antibody-enhanced polymer methodCancer20121184427443622252991
- ChristensenJGZouHYArangoMECytoreductive antitumor activity of PF-2341066, a novel inhibitor of anaplastic lymphoma kinase and c-Met, in experimental models of anaplastic large-cell lymphomaMol Cancer Ther200763314332218089725
- ZouHYLiQLeeJHAn orally available small-molecule inhibitor of c-Met, PF-2341066, exhibits cytoreductive antitumor efficacy through antiproliferative and antiangiogenic mechanismsCancer Res2007674408441717483355
- CamidgeDRBangYJKwakELActivity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a phase 1studyLancet Oncol2012131011101922954507
- ShawATKimDWNakagawaKPhase III study of crizotinib versus pemetrexed or docetaxel chemotherapy in patients with advanced ALK-positive non-small cell lung cancer (NSCLC) (PROFILE 1007)Ann Oncol201223Suppl 9ixe21
- KimDWAhnMJShiYUpdated results of a global phase II study with crizotinib in advanced ALK-positive non-small cell lung cancerAnn Oncol201223Suppl 9ix400ix446
- SakamotoHTsukaguchiTHiroshimaSCH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutantCancer Cell20111967969021575866
- KiuraKSetoTYamamotoNA first-in-human phase I/II study of ALK inhibitor CH5424802 in patients with ALK-positive NSCLCJ Clin Oncol201230Suppl7602
- NishioMKiuraKNakagawaKA phase I/II study of ALK inhibitor CH5424802 in patients with ALK-positive NSCLC; safety and efficacy interim results of the phase II portionAnn Oncol201223Suppl 9ix152ix174
- ShawATCamidgeDRFelipEResults of a first-in-human phase I study of the ALK INHIBITOR LDK378 in advanced solid tumorsAnn Oncol201223Suppl 9ix152ix174
- CecconMMologniLBissonWScapozzaLGambacorti-PasseriniCCrizotinib-resistant NPM-ALK mutants confer differential sensitivity to unrelated Alk inhibitorsMol Cancer Res20121112213223239810
- KatayamaRKhanTMBenesCTherapeutic strategies to overcome crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALKProc Natl Acad Sci USA20111087535754021502504
- GettingerSWeissGJSalgiaRA first-in-human dose-finding study of the ALK/EGFR inhibitor AP26113 in patients with advanced malignanciesAnn Oncol201223Suppl 9ix152ix174
- LovlyCMHeuckmannJMde StanchinaEInsights into ALK-driven cancers revealed through development of novel ALK tyrosine kinase inhibitorsCancer Res2011714920493121613408
- KuromitsuSMoriMShimadaIAntitumor activities of ASP3026 against EML4-ALK-dependent tumor modelsMol Cancer Ther201110SupplA227