Abstract
Angiogenesis has long been recognized as an essential element in tumor growth. Since the conception of antiangiogenesis for cancer therapeutics, great strides have been made in understanding the molecular biology underlying angiogenesis, both in cancer and in physiology. By capitalizing on these advancements through bench-to-bedside research, potent antiangiogenic agents have been developed and tested. To date, the clinical results of most of these antiangiogenic agents have not met expectations. Even with the most successful agents, such as bevacizumab, used either as single agents or in combination with chemotherapy, gains in overall survival of cancer patients have been modest in most cases. In this article, the authors present the evolving views of antiangiogenic therapy, review recent experimental and clinical studies on antiangiogenesis, and address the fundamental role of hypoxia in tumor progression, which may be key to improving the efficacy of antiangiogenic therapy.
Angiogenesis, tumor growth, and antiangiogenesis
Recognition of the association between vigorous neovascularization (angiogenesis) and tumor growth dates back to the first half of the 20th century.Citation1,Citation2 However, the significance of this finding and the relationship between new vessel growth and tumor growth were not fully appreciated until many years later. Folkman and others first showed that, in the absence of vascularization, tumor growth arrested when the tumor reached 2–3 mm in diameter, presumably owing to limited diffusion of oxygen, nutrients, and waste products. Furthermore, small, dormant tumors would quickly resume expansion when allowed to initiate neovascularization.Citation3–Citation5
An angiogenic factor was later isolated from human and animal tumors that is mitogenic to endothelial cells and stimulates the rapid formation of new capillaries in animals.Citation6 Folkman proposed targeting this angiogenic factor for angiogenic inhibition as a strategy for cancer therapy, which is the fundamental premise of antiangiogenesis therapy. This unconventional approach seemed promising for several reasons. First, fewer side effects were expected from inhibiting angiogenesis than from traditional cytotoxic agents, presumably because new vessel growth in an adult patient is less important under most physiologic conditions (eg, in the absence of wound healing). Second, it seemed plausible that, in addition to restricting tumor growth to the limits of direct diffusion of nutrients and waste metabolites, reduced access to the vasculature would decrease distant metastasis. Third, by targeting the vasculature, one would expect less likelihood of developing resistance from endothelial cells than from tumor cells.Citation7,Citation8
Identifying this angiogenic factor, however, proved to be challenging. In time, it became apparent that there were many different factors involved in stimulating angiogenesis. The first to be identified was the basic fibroblast growth factor (bFGF),Citation9 which was shown to directly stimulate endothelial cell proliferation.Citation10 A few years later, vascular endothelial growth factor (VEGF) was identified, cloned,Citation11,Citation12 and characterized as a potent regulator of angiogenesis via mitogenic and antiapoptotic signaling in endothelial cells.Citation13,Citation14 As an increasing number of angiogenic regulators were identified and characterized, including the angiopoietins,Citation15,Citation16 interleukin 8,Citation17,Citation18 and others, a more cohesive view of the mechanisms of angiogenesis in tumor growth developed.
Angiogenic switch
In 1991, Hanahan and colleagues further developed the theory that angiogenesis is required for continued tumor growth by demonstrating a switch from low to high vessel density during the multistep process of fibrosarcoma progression in transgenic mice.Citation19 It was found that cells cultured from advanced preneoplastic lesions secreted bFGF into the growth medium, whereas cells derived from lower-grade lesions did not. This ‘angiogenic switch’ correlated not only with histologically higher-grade tumors but also with tumorigenicity. Similar results seen in islet cell carcinoma and epidermal squamous cell carcinoma mouse models added additional evidence to the idea that the switch to an angiogenic phenotype is a discrete step during tumor progression and essential for solid tumor growth.Citation20
The importance of tumor angiogenesis became more apparent as the underlying mechanisms began to come into focus. Folkman’s vision of developing antiangiogenic cancer therapies also began to seem within reach as interest increased. This step forward, however, did not come without hindrance.
Setbacks for antiangiogenic therapy
Despite the promising preclinical and animal model data, almost none of the early agents identified and tested have made it past clinical trials. A new use for thalidomide, which was originally introduced as a sedative and antiemetic drug, was postulated after its metabolites were found to inhibit angiogenesis in a rabbit cornea micropocket assay.Citation21 Unfortunately, early trials showed only modest clinical effectiveness in prostate cancer patients and no antitumor activity in recurrent or metastatic squamous cell carcinoma of the head and neck.Citation22,Citation23
Trials of a host of other antiangiogenic drugs followed with similar results. TNP-470, an analog of fumigillin, generated only one short-lived partial response in 33 patients with metastatic renal carcinoma,Citation24 and a later study also failed to show clinical benefit for prostate cancer.Citation25 In two phase I clinical trials, angiostatin, which is a naturally occurring angiogenesis inhibitor,Citation26 showed no clinical response with several types of solid tumors.Citation27,Citation28 Likewise, endostatin, another natural angiogenesis inhibitor,Citation29 showed only minor antitumor activity in a phase I trialCitation30 and no significant tumor regression in patients with advanced neuroendocrine tumors in a phase II trial.Citation31 In a phase II clinical trial of ABT-510, which is a peptide mimetic of thrombospondin type 1 (yet another endogenous angiogenesis inhibitor), only 3 out of 21 late-stage malignant melanoma patients showed stable disease, and no definite clinical efficacy was demonstrated.Citation32 Another phase II trial also showed only one objective response out of 88 patients with advanced soft tissue sarcoma.Citation33
The mechanisms of action for all the previously discussed drugs are poorly or only partially understood. Agents with specific molecular targets in the angiogenic signaling pathways were explored as alternatives. This approach, however, also had several disappointments before any measure of clinical success was achieved. SU5416, which is a small synthetic receptor tyrosine kinase inhibitor of the VEGF receptor VEGFR-2, produced no objective response in 27 patients with refractory multiple myeloma. Two other phase II trials showed either no or rare responses in patients with advanced soft tissue sarcomas and recurrent head and neck cancers.Citation34 PTK787/ZK 222584, which is another small molecule tyrosine kinase receptor inhibitor targeting all of the VEGF receptors, also failed to produce significant responses in patients with acute myeloid leukemia.Citation35
Signs of success
In 2003, an anti-VEGF humanized monoclonal antibody, bevacizumab, made its debut with only slightly better results as a monotherapy in metastatic renal cancer patients;Citation36 when compared with a placebo, high-dose bevacizumab had only a 10% response rate with modestly prolonged progression-free survival but no overall survival benefit. Although these initial results were disappointing, evidence of the efficacy of bevacizumab came shortly thereafter, leading to an eventual paradigm shift in the concept of antiangiogenic therapy. In June 2004, Hurwitz et al reported a phase III trial showing substantial improvement in overall survival (about a 5-month increase) in patients with metastatic colorectal cancer when treated with bevacizumab combined with irinotecan, fluorouracil, and leucovorin.Citation37 Although a handful of phase III trials involving treatment of different cancers by combining bevacizumab with various regimens have shown improvement only in progression-free survival but not overall survival,Citation38–Citation40 most reported studies have shown increased overall survival along with increased progression-free survival when bevacizumab is added to the treatment regimen. These include a 2-month overall survival benefit in patients with metastatic renal cell carcinoma when treated with bevacizumab in combination with interferon α-2a;Citation41 in patients who had previously been treated for metastatic colorectal cancer when combined with oxaliplatin, fluorouracil, and leucovorin;Citation42 and in patients with non-small-cell lung cancer when combined with paclitaxel and carboplatin.Citation43 A 3-month overall survival benefit was also reported in patients with metastatic colorectal cancer treated with bevacizumab in combination with fluorouracil and leucovorin.Citation44 Therefore, in most cases, an apparent synergy is seen in which bevacizumab has clinical benefit when combined with cytotoxic chemotherapies.
It is also interesting to note that thalidomide, as discussed earlier, has not been proven effective as a monotherapy, but, when combined with melphalan, a cytotoxic alkylating agent, and prednisone, an immunosuppressing corticosteroid, it increases median overall survival by nearly 18 months in elderly patients with multiple myeloma,Citation45 possibly by reducing bone marrow vascularization.Citation46 Still, even with these first signs of success involving bevacizumab and thalidomide, tumors did become resistant relatively quickly and overall improvement was modest.
Normalization of tumor vasculature
With these clinical data has come a paradox. If antiangiogenic therapy destroys tumor vasculature, a reduction of drug delivery would be expected. Why then does antiangiogenesis enhance tumor killing when combined with cytotoxic agents? Perhaps even more puzzling is the observation that anti-VEGF therapy can increase tumor irradiation efficacy,Citation47 which is largely dependent on tissue oxygenation.
To account for these apparently conflicting findings, Jain posited that, in addition to destroying vasculature for depriving the tumor of oxygen and nutrients, antiangiogenic agents also transiently ‘normalize’ the abnormal structure and function of tumor vasculature to make it more efficient for oxygen and drug delivery.Citation48 Vasculature maintenance involves a homeostatic interplay between pro- and antiangiogenic signals. In normal tissue these signals are balanced, but during neoplastic growth the proangiogenic signals are over-expressed, thereby stimulating inappropriate vessel growth and leading to characteristically disorganized, inefficient, and leaky tumor vasculature. Jain proposed that using a low dose or ‘judicious’ application of antiangiogenic agents could restore the balance in the angiogenic regulation by pruning immature, nonproductive vessels, decreasing vessel permeability, and reducing abnormal dilation. The expected functional consequence of vasculature normalization is decreased interstitial fluid pressure, relieved hypoxic stress, and improved penetration of drugs in the tumor.
Although vasculature normalization has been shown to improve vascular function in preclinical models through intra-vital imaging studies,Citation49 it is not straightforward to ascertain in patients the changes in blood flow and distribution within a tumor during antiangiogenic therapy, making it difficult to verify this process. Many questions, such as why there is only a transient ‘normalization window’ and how to identify it, remain unanswered.
Negative sequelae of targeting tumor vasculature
As mentioned earlier, a potential advantage of targeting endothelial rather than tumor cells is the avoidance of drug resistance, because endothelial cells, unlike those of tumors, are genetically stable.Citation7 However, tumor revascularization following a transient decrease in vessel density with an anti-VEGF receptor agent has been reported, resulting at least in part from increased levels of bFGF.Citation50
It has become increasingly clear that VEGF-targeted therapy probably involves multiple mechanisms.Citation51 A possible explanation for the activation of alternative angiogenic pathways, however, is that antiangiogenesis induces hypoxia, resulting in activation of the hypoxia-inducible factor α (HIF-α), which in turn triggers revascularization of the tumor. HIF-α is known to transcriptionally upregulate a host of pro- and antiangiogenic genes encoding placental growth factor, angiopoietin 1, angiopoietin 2, stromal-derived factor 1, platelet-derived growth factor, and bFGF, as well as VEGF.Citation52 Given that overexpression of HIF-1α, one of the HIF-α family members (see below), induces nonleaky hypervascularity in transgenic mice,Citation53 it is conceivable that activated HIF-α could upregulate both pro- and antiangiogenic factors for neovascularization in tumors. Indeed, mice haplodeficient in Egln1, which encodes a negative regulator of HIF-α (see below), showed normalized endothelial lining and vessel maturation, thereby resulting in improved tumor oxygenation with decreased tumor invasiveness, even though tumor growth was not inhibited.Citation54
Irrespective of vasculature regression and/or normalization, a more serious concern with antiangiogenic therapy is that vessel regression may drive tumors toward a more locally invasive and distantly metastatic phenotype. Indeed, increased invasiveness in glioblastoma models was observed after systemic antiangiogenic therapy with an antibody against VEGFR-2, despite a marked inhibition of tumor growth and microvessel density.Citation55,Citation56 Similar results were seen after treatment with bevacizumab in another glioblastoma preclinical study.Citation57 In addition, increased invasiveness after anti-VEGFR-2 treatment was also seen in a pancreatic neuroendocrine cancer mouse model, along with increased liver and lymph node metastases.Citation58 Furthermore, short-term treatment with a potent angiogenic inhibitor, sunitinib/SU11248, also accelerated metastasis into multiple organs in a preclinical study using breast cancer and melanoma cells.Citation59 More troubling is that the invasive nature of the tumors seemed to be permanently established, as removal of treatment did not relieve the aggressive phenotype, possibly suggesting a genetic transformation. Although this increased invasiveness in response to antiangiogenic therapy has not been unequivocally confirmed in patients, partially because of imaging limitations in evaluating these tumors (ie, FLAIR magnetic resonance imaging), it has nonetheless been observed at least subjectively.Citation60 A review of literature indicates that although glioblastoma patients benefit greatly from reduced cerebral edema and intracranial pressure through angiogenic inhibition, tumor invasion continues.Citation61 Additional concerns include that the normalization of tumor vasculature by antiangiogenic agents may restore the blood–brain barrier function, thereby antagonizing the efficacy of chemotherapeutic drugs.
HIF-1α, genetic alteration, and tumor progression
Hypoxia has long been implicated in genetic instability and tumor progression, which may account for the inevitable failure of antiangiogenesis as a monotherapy. Although the mechanisms underlying hypoxia-induced tumor progression remain to be elucidated, recent evidence indicates that HIF-α plays an essential role in tumor growth and progression.
In human cancers, both HIF-1α and HIF-2α, two of the prevalent members of the HIF-α family, are frequently dysregulated, resulting in their overexpression.Citation62 Under physiological conditions when oxygen tension within cells is high, HIF-α is hydroxylated at specific proline residues by the prolyl hydroxylases EGLN1, EGLN2, and EGLN3 (better known as PHD2, PHD1, and PHD3, respectively).Citation63,Citation64 This allows HIF-α to be ubiquitinated by a pVHL-directed E3 ligase and targeted to the proteasome for degradation. In hypoxia, however, the hydroxylation reaction is inhibited; HIF-α accumulates within the cell, translocates to the nucleus, and, upon dimerization with its binding partner aryl hydrocarbon receptor nuclear translocator (ARNT), acts as a transcription factor for the activation of a diverse group of hypoxia-responsive genes.Citation52 This canonical HIF-α–ARNT pathway () has accounted for hypoxic activation of many genes directly related to tumor growth and survival, such as those involved in glycolysis, cell migration, apoptosis, multidrug resistance, extracellular matrix modification, epithelial–mesenchymal transition, and angiogenesis.Citation62,Citation65
Figure 1 A schematic representation of the HIF-1α–ARNT pathway and the HIF-1α–c-Myc pathway. Stabilized HIF-1α participates in the canonical HIF-1α– ARNT pathway (–ARNT) through dimerization with its binding partner ARNT, recruitment of the transcription coactivator p300/CBP, and binding to the HRE in the promoter of the angiogenic and glycolytic genes for transcriptional activation. Alternatively, the HIF-1α–c-Myc pathway (–c-Myc) involves HIF-1α competing with c-Myc for binding to the transcription factor Sp1 in the promoter of DNA repair genes, resulting in selective c-Myc displacement and gene repression.
Abbreviations: HIF, hypoxia-inducible factor; ARNT, aryl hydrocarbon receptor nuclear translocator; HRE, hypoxia-responsive element.
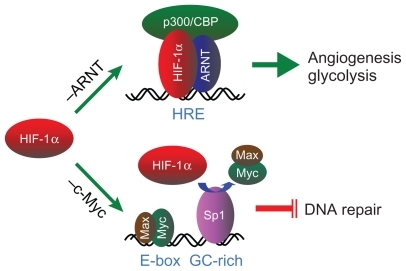
Despite these extraordinary insights into the mechanisms underlying tumor biology, how hypoxia drives genetic alteration, the underlying cause of tumor progression, has yet to be elucidated.Citation66,Citation67 Interestingly, we and others began to show recently that HIF-1α and HIF-2α have opposing effects on DNA repair; HIF-1α inhibits, whereas HIF-2α stimulates, DNA repair.Citation68,Citation69 Although how these conflicting effects between HIF-1α and HIF-2α are reconciled within tumor cells needs further investigation, our results demonstrated that HIF-1α, but not HIF-2α, is essential to hypoxic downregulation of the DNA mismatch repair genes MSH2 and MSH6 Citation70 and the double-strand break repair gene NBN.Citation71 Interestingly, HIF-1α does so by a distinct mechanism that is independent of the HIF-1α–ARNT pathway but involves HIF-1α functionally counteracting c-Myc, a transcriptional activator for maintaining DNA repair gene expression. This HIF-1α–c-Myc pathwayCitation72 accounts not only for hypoxic inhibition of DNA repair but also for resultant DNA damage and genetic alterations (). By uncoupling these two distinct, independent pathways of HIF-1α, we have recently shown that the HIF-1α–c-Myc pathway is essential to drive tumor progression, whereas the HIF-1α–ARNT pathway is more involved in tumor growth.Citation73 Therefore, the dual functions of HIF-1α may account on the one hand for vasculature normalization resulting from regulated expression of both pro- and antiangiogenic genes via the HIF-1α–ARNT pathway, and on the other hand for tumor progression driven by genetic alterations via the HIF-1α–c-Myc pathway.
With this gained knowledge, we propose that in addition to its important role in angiogenesis and glycolysis for tumor growth and survival, HIF-1α is essential to drive genetic alteration for tumor progression, which is a negative aspect of the hypoxic responseCitation74 enabling tumor cells to evolve through increased genetic heterogeneity. This could explain the ease with which many cancers are able to adapt to a wide variety of therapeutics (including antiangiogenics) and develop resistance. It could also explain the apparent genetic changes that lead to increased invasion and metastasis in antiangiogenic-treated tumors.
Future directions of antiangiogenic therapy
Although antiangiogenic therapy remains promising,Citation51 a durable antitumor activity for an improved overall survival is desired. To this end, several hypotheses have been proposed. Pietras and Hanahan suggested the use of broader-spectrum angiogenesis inhibitors or ‘cocktails’ of specific inhibitors as a method of blocking alternative angiogenic pathways that may be activated under a VEGF blockade.Citation75 They have demonstrated the efficacy of this tactic in an animal model of islet cell carcinogenesis. Treatment with anti-VEGFR-2 antibodies led to an initial decrease in tumor vascularity as well as tumor size. This was followed by revascularization and regrowth of the tumors. Greater response was seen, however, by coinhibiting bFGF, which was suspected in an alternative angiogenic pathway. This resulted in a further decrease in tumor growth after the initial regression.
On the other hand, it stands to reason that if HIF-α can be targeted alongside antiangiogenic agents to prevent the induction of genetic alteration and/or angiogenesis, this could greatly improve the efficacy of antiangiogenic therapy. Interestingly, Melillo and Rapisarda et al have identified a potential HIF-α inhibitor, topotecan.Citation76,Citation77 When used alongside bevacizumab in U251 glioma xenografts, topotecan showed considerable synergistic antitumor activity. Not only was tumor volume decreased but intratumor vasculature was also decreased compared with tumors treated with either topotecan or bevacizumab alone.Citation78 Considering the increased invasive nature of tumors following antiangiogenic treatment, HIF-α targeting may prove to be an effective way of maximizing antiangiogenic therapy in the future. Likewise, drugs that potentially block genetic alteration and thereby tumor progression may greatly improve overall survival when combined with antiangiogenic agents.
Conclusions
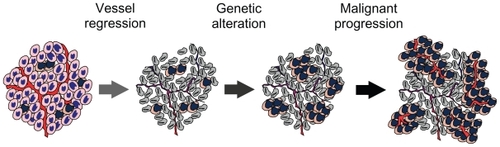
Antiangiogenic therapy was initially based on the notion that angiogenesis is required for tumor growth, and thus destruction of the tumor vasculature would deprive the tumor of oxygen and nutrients, resulting in growth inhibition. However, tumor vasculature is structurally abnormal and functionally inefficient, and the resultant hypoxic microenvironment is associated with tumor progression and resistance to therapies (). Therefore, therapeutic destruction of the tumor vasculature is expected to yield more severe hypoxia, which on the one hand induces additional angiogenic responses through the activation of HIF-α for normalizing vasculature, and on the other hand drives genetic alteration for malignant progression. This view accounts for the unexpected clinical outcomes when single antiangiogenesis agents are used, some clinical benefits when antiangiogenesis agents are used in combination with chemotherapy, and fundamentally the inevitable problem of angiogenic therapy, which is that hypoxia promotes tumor progression. Thus, targeting tumor hypoxia may improve the efficacy of angiogenic therapy.
Figure 2 A hypothetic model illustrates that malignant progression results from antiangiogenic therapy. Angiogenic inhibition deprives tumor cells of oxygen and nutrients, resulting in vessel regression and thereby death of the majority of the tumor cells. However, hypoxic cells harbored within the solid tumor are able to tolerate severe hypoxia by undergoing genetic alterations for malignant progression via the HIF-1α–c-Myc pathway and by inducing angiogenesis and glycolysis for cell proliferation via the HIF-1α–ARNT pathway.
Abbreviations: HIF, hypoxia-inducible factor; ARNT, aryl hydrocarbon receptor nuclear translocator.
Acknowledgments
We thank Kristin Kraus for editing the manuscript. LEH was supported in part by a Public Health Service grant (CA-131355) from the National Cancer Institute, and CR was supported by a Public Health Service grant (HL-007744) from the National Institute of Diabetes and Digestive and Kidney Diseases.
Disclosure
The authors report no conflicts of interest in this work.
References
- IdeAGBakerNHWarrenSLVascularization of the Brown–Pearce rabbit epithelioma transplant as seen in the transparent ear chamberAJR Am J Roentgenol193942891899
- AlgireGHChalkleyHWVascular reactions of normal and malignant tissues in vivo. I. Vascular reactions of mice to wounds and to normal and neoplastic transplantsJ Natl Cancer Inst194567385
- GreeneHSHeterologous transplantation of mammalian tumors: I. The transfer of rabbit tumors to alien speciesJ Exp Med194173446147419871090
- FolkmanJColePZimmermanSTumor behavior in isolated perfused organs: in vitro growth and metastases of biopsy material in rabbit thyroid and canine intestinal segmentAnn Surg196616434915025951515
- GimbroneMAAsterRHCotranRSCorkeryJJandlJHFolkmanJPreservation of vascular integrity in organs perfused in vitro with a platelet-rich mediumNature1969222518833365775827
- FolkmanJTumor angiogenesis: therapeutic implicationsN Engl J Med197128521118211864938153
- BoehmTFolkmanJBrowderTO’ReillyMSAntiangiogenic therapy of experimental cancer does not induce acquired drug resistanceNature199739066584044079389480
- FolkmanJSeminars in medicine of the Beth Israel Hospital, Boston. Clinical applications of research on angiogenesisN Engl J Med199533326175717637491141
- ShingYFolkmanJSullivanRButterfieldCMurrayJKlagsbrunMHeparin affinity: purification of a tumor-derived capillary endothelial cell growth factorScience19842234642129612996199844
- FolkmanJKlagsbrunMAngiogenic factorsScience198723547874424472432664
- KeckPJHauserSDKriviGVascular permeability factor, an endothelial cell mitogen related to PDGFScience19892464935130913122479987
- LeungDWCachianesGKuangWJGoeddelDVFerraraNVascular endothelial growth factor is a secreted angiogenic mitogenScience19892464935130613092479986
- AlonTHemoIItinAPe’erJStoneJKeshetEVascular endothelial growth factor acts as a survival factor for newly formed retinal vessels and has implications for retinopathy of prematurityNat Med1995110102410287489357
- FerraraNDavis-SmythTThe biology of vascular endothelial growth factorEndocr Rev19971814259034784
- DavisSAldrichTHJonesPFIsolation of angiopoietin-1, a ligand for the TIE2 receptor, by secretion-trap expression cloningCell1996877116111698980223
- HolashJMaisonpierrePCComptonDVessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGFScience199928454221994199810373119
- KochAEPolveriniPJKunkelSLInterleukin-8 as a macro-phage- derived mediator of angiogenesisScience19922585089179818011281554
- StrieterRMKunkelSLElnerVMInterleukin-8. A corneal factor that induces neovascularizationAm J Pathol19921416127912841281615
- KandelJBossy-WetzelERadvanyiFKlagsbrunMFolkmanJHanahanDNeovascularization is associated with a switch to the export of bFGF in the multistep development of fibrosarcomaCell1991666109511041717155
- HanahanDFolkmanJPatterns and emerging mechanisms of the angiogenic switch during tumorigenesisCell19968633533648756718
- D’AmatoRJLoughnanMSFlynnEFolkmanJThalidomide is an inhibitor of angiogenesisProc Natl Acad Sci U S A1994919408240857513432
- TsengJEGlissonBSKhuriFRPhase II study of the antiangiogenesis agent thalidomide in recurrent or metastatic squamous cell carcinoma of the head and neckCancer20019292364237311745292
- FiggWDDahutWDurayPA randomized phase II trial of thalidomide, an angiogenesis inhibitor, in patients with androgenin-dependent prostate cancerClin Cancer Res2001771888189311448901
- StadlerWMKuzelTShapiroCSosmanJClarkJVogelzangNJMulti-institutional study of the angiogenesis inhibitor TNP-470 in metastatic renal carcinomaJ Clin Oncol19991782541254510561320
- LogothetisCJWuKKFinnLDPhase I trial of the angiogenesis inhibitor TNP-470 for progressive androgen-independent prostate cancerClin Cancer Res2001751198120311350884
- O’ReillyMSHolmgrenLShingYAngiostatin: a novel angiogenesis inhibitor that mediates the suppression of metastases by a Lewis lung carcinomaCell19947923153287525077
- BeerepootLVWitteveenEOGroenewegenGRecombinant human angiostatin by twice-daily subcutaneous injection in advanced cancer: a pharmacokinetic and long-term safety studyClin Cancer Res20039114025403314519623
- SoffGAWangHCundiffDLIn vivo generation of angiostatin isoforms by administration of a plasminogen activator and a free sulfhydryl donor: a phase I study of an angiostatic cocktail of tissue plasminogen activator and mesnaClin Cancer Res200511176218622516144924
- O’ReillyMSBoehmTShingYEndostatin: an endogenous inhibitor of angiogenesis and tumor growthCell19978822772859008168
- HerbstRSHessKRTranHTPhase I study of recombinant human endostatin in patients with advanced solid tumorsJ Clin Oncol200220183792380312228199
- KulkeMHBergslandEKRyanDPPhase II study of recombinant human endostatin in patients with advanced neuroendocrine tumorsJ Clin Oncol200624223555356116877721
- MarkovicSNSumanVJRaoRAA phase II study of ABT-510 (thrombospondin-1 analog) for the treatment of metastatic melanomaAm J Clin Oncol200730330330917551310
- BakerLHRowinskyEKMendelsonDRandomized, phase II study of the thrombospondin-1-mimetic angiogenesis inhibitor ABT-510 in patients with advanced soft tissue sarcomaJ Clin Oncol200826345583558818981463
- FuryMGZahalskyAWongRA phase II study of SU5416 in patients with advanced or recurrent head and neck cancersInvest New Drugs200725216517216983506
- RobozGJGilesFJListAFPhase 1study of PTK787/ZK 222584, a small molecule tyrosine kinase receptor inhibitor, for the treatment of acute myeloid leukemia and myelodysplastic syndromeLeukemia200620695295716617323
- YangJCHaworthLSherryRMA randomized trial of bevacizumab, an anti-vascular endothelial growth factor antibody, for metastatic renal cancerN Engl J Med2003349542743412890841
- HurwitzHFehrenbacherLNovotnyWBevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancerN Engl J Med2004350232335234215175435
- van CutsemEVervenneWLBennounaJPhase III trial of bevacizumab in combination with gemcitabine and erlotinib in patients with metastatic pancreatic cancerJ Clin Oncol200927132231223719307500
- SaltzLBClarkeSDiaz-RubioEBevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: a randomized phase III studyJ Clin Oncol200826122013201918421054
- MillerKWangMGralowJPaclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancerN Engl J Med2007357262666267618160686
- EscudierBBellmuntJNegrierSPhase III trial of bevacizumab plus interferon alfa-2a in patients with metastatic renal cell carcinoma (AVOREN): final analysis of overall survivalJ Clin Oncol201028132144215020368553
- GiantonioBJCatalanoPJMeropolNJBevacizumab in combination with oxaliplatin, fluorouracil, and leucovorin (FOLFOX4) for previously treated metastatic colorectal cancer: results from the Eastern Cooperative Oncology Group Study E3200J Clin Oncol200725121539154417442997
- SandlerAGrayRPerryMCPaclitaxel–carboplatin alone or with bevacizumab for non-small-cell lung cancerN Engl J Med2006355242542255017167137
- KabbinavarFFHambletonJMassRDHurwitzHIBergslandESarkarSCombined analysis of efficacy: the addition of bevacizumab to fluorouracil/leucovorin improves survival for patients with metastatic colorectal cancerJ Clin Oncol200523163706371215867200
- FaconTMaryJYHulinCMelphalan and prednisone plus thalidomide versus melphalan and prednisone alone or reduced-intensity autologous stem cell transplantation in elderly patients with multiple myeloma (IFM 99-06): a randomised trialLancet200737095941209121817920916
- SinghalSMehtaJDesikanRAntitumor activity of thalidomide in refractory multiple myelomaN Engl J Med1999341211565157110564685
- LeeCGHeijnMdi TomasoEAnti-vascular endothelial growth factor treatment augments tumor radiation response under normoxic or hypoxic conditionsCancer Res200060195565557011034104
- JainRKNormalization of tumor vasculature: an emerging concept in antiangiogenic therapyScience20053075706586215637262
- FukumuraDDudaDGMunnLLJainRKTumor microvasculature and microenvironment: novel insights through intravital imaging in pre-clinical modelsMicrocirculation201017320622520374484
- CasanovasOHicklinDJBergersGHanahanDDrug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumorsCancer Cell20058429930916226705
- EllisLMHicklinDJVEGF-targeted therapy: mechanisms of anti-tumour activityNat Rev Cancer20088857959118596824
- SemenzaGLTargeting HIF-1 for cancer therapyNat Rev Cancer200331072173213130303
- ElsonDAThurstonGHuangLEInduction of hypervascularity without leakage or inflammation in transgenic mice overexpressing hypoxia-inducible factor-1alphaGenes Dev200115192520253211581158
- MazzoneMDettoriDLeite de OliveiraRHeterozygous deficiency of PHD2 restores tumor oxygenation and inhibits metastasis via endothelial normalizationCell2009136583985119217150
- LamszusKKunkelPWestphalMInvasion as limitation to antiangiogenic glioma therapyActa Neurochir Suppl20038816917714531575
- BergersGHanahanDModes of resistance to anti-angiogenic therapyNat Rev Cancer20088859260318650835
- Lucio-EterovicAKPiaoYde GrootJFMediators of glioblastoma resistance and invasion during antivascular endothelial growth factor therapyClin Cancer Res200915144589459919567589
- Paez-RibesMAllenEHudockJAntiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasisCancer Cell200915322023119249680
- EbosJMLeeCRCruz-MunozWBjarnasonGAChristensenJGKerbelRSAccelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesisCancer Cell200915323223919249681
- NordenADYoungGSSetayeshKBevacizumab for recurrent malignant gliomas: efficacy, toxicity, and patterns of recurrenceNeurology2008701077978718316689
- VerhoeffJJvan TellingenOClaesAConcerns about antiangiogenic treatment in patients with glioblastoma multiformeBMC Cancer2009944420015387
- BertoutJAPatelSASimonMCThe impact of O2 availability on human cancerNat Rev Cancer200881296797518987634
- HuangLEBunnHFHypoxia-inducible factor and its biomedical relevanceJ Biol Chem200327822195751957812639949
- KaelinWGJrRatcliffePJOxygen sensing by metazoans: the central role of the HIF hydroxylase pathwayMol Cell200830439340218498744
- HarrisALHypoxia – a key regulatory factor in tumour growthNat Rev Cancer200221384711902584
- HuangLEBindraRSGlazerPMHarrisALHypoxia-induced genetic instability – a calculated mechanism underlying tumor progressionJ Mol Med200785213914817180667
- BristowRGHillRPHypoxia and metabolism. Hypoxia, DNA repair and genetic instabilityNat Rev Cancer20088318019218273037
- HuangLECarrot and stick: HIF-alpha engages c-Myc in hypoxic adaptationCell Death Differ200841567267718188166
- GordanJDLalPDondetiVRHIF-alpha effects on c-Myc distinguish two subtypes of sporadic VHL-deficient clear cell renal carcinomaCancer Cell200814643544619061835
- KoshijiMToKKHammerSHIF-1alpha induces genetic instability by transcriptionally downregulating MutSalpha expressionMol Cell200517679380315780936
- ToKKSedelnikovaOASamonsMBonnerWMHuangLEThe phosphorylation status of PAS-B distinguishes HIF-1alpha from HIF-2alpha in NBS1 repressionEMBO J200625204784479417024177
- KoshijiMKageyamaYPeteEAHorikawaIBarrettJCHuangLEHIF-1alpha induces cell cycle arrest by functionally counteracting MycEMBO J20042391949195615071503
- YooYGChristensenJHuangLEHIF-1α confers aggressive malignant traits on human tumor cells independent of its canonical transcriptional functionCancer Res
- ToKKKoshijiMHammerSHuangLEGenetic instability: the dark side of the hypoxic responseCell Cycle20054788188215970707
- PietrasKHanahanDA multitargeted, metronomic, and maximum-tolerated dose ‘chemo-switch’ regimen is antiangiogenic, producing objective responses and survival benefit in a mouse model of cancerJ Clin Oncol200523593995215557593
- MelilloGInhibiting hypoxia-inducible factor 1 for cancer therapyMol Cancer Res20064960160516940159
- RapisardaAUranchimegBSordetOPommierYShoemakerRHMelilloGTopoisomerase I-mediated inhibition of hypoxia-inducible factor 1: mechanism and therapeutic implicationsCancer Res20046441475148214983893
- RapisardaAHollingsheadMUranchimegBIncreased antitumor activity of bevacizumab in combination with hypoxia inducible factor-1 inhibitionMol Cancer Ther2009871867187719584228