Abstract
Patients with chronic obstructive pulmonary disease (COPD) exhibit dominant features of chronic bronchitis, emphysema, and/or asthma, with a common phenotype of airflow obstruction. COPD pulmonary physiology reflects the sum of pathological changes in COPD, which can occur in large central airways, small peripheral airways, and the lung parenchyma. Quantitative or high-resolution computed tomography is used as a surrogate measure for assessment of disease progression. Different biological or molecular markers have been reported that reflect the mechanistic or pathogenic triad of inflammation, proteases, and oxidants and correspond to the different aspects of COPD histopathology. Similar to the pathogenic triad markers, genetic variations or polymorphisms have also been linked to COPD-associated inflammation, protease–antiprotease imbalance, and oxidative stress. Furthermore, in recent years, there have been reports identifying aging-associated mechanistic markers as downstream consequences of the pathogenic triad in the lungs from COPD patients. For this review, the authors have limited their discussion to a review of mechanistic markers and genetic variations and their association with COPD histopathology and disease status.
Introduction
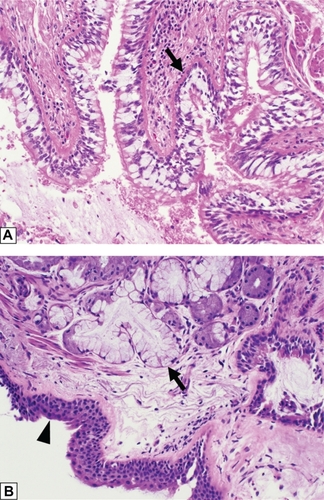
Chronic obstructive pulmonary disease (COPD) manifests in two clinical phenotypes, bronchitis and emphysema.Citation1,Citation2 Chronic bronchitis is clinically defined as a productive cough on most days of a month for at least 3 months of a year in 2 or more consecutive years.Citation3 Although cigarette smoking is the major cause for chronic bronchitis, other causes include air pollution and occupational exposures. These exposures introduce particulates and oxidants resulting in a common pathogenesis.Citation2 Thus, the gross pathology and histology of chronic bronchitis is the same regardless of the etiology. Gross examination of lung tissue in a patient with chronic bronchitis shows thickened bronchial walls with luminal narrowing, and mucous plugging or mucopurulent debris within the airways. Microscopically, these gross findings correspond to goblet cell hyperplasia, thickening of the subepithelial basement membrane, bronchial wall fibrosis, and hyperplasia of the subepithelial seromucinous glands; classically defined by the Reid index ().Citation4 A chronic inflammatory infiltrate may also be present, but is not required, and squamous metaplasia of the respiratory epithelium can also be seen.
Figure 1 Histologic features of chronic bronchitis. (A) A section of bronchiole wall with luminal accumulation of mucous, goblet cell hyperplasia, basement membrane thickening (arrow), and scattered mononuclear inflammatory cells. (B) A bronchial wall with squamous metaplasia of the luminal epithelium (arrow head) and hyperplasia of the subepithelial seromucinous glands (arrow).
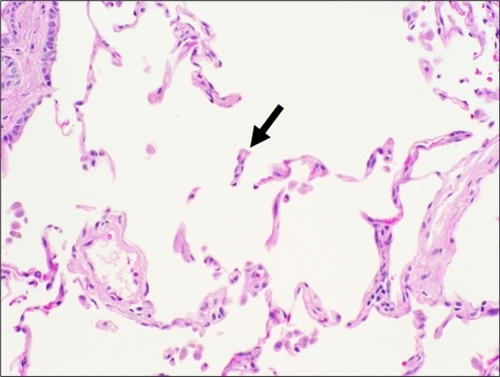
There are several types of emphysema that share in common destruction of alveolar walls with resultant enlargement of airspaces distal to terminal bronchioles. For the purposes of this discussion, the Pratt classificationCitation5 will be used with the Thurlbeck classificationCitation6 given in parentheses. Centrilobular (centriacinar) emphysema is most commonly caused by cigarette smoking and is defined by destruction of alveoli centered around the respiratory bronchiole and involving the proximal acinus.Citation7 Gross examination shows punctuate areas of small airspace destruction, often associated with the deposition of pigment, which is typically more pronounced in the apices. Panlobular (panacinar) emphysema is caused by α1-antitrypsin deficiency (A1ATD); an inherited disorder involving chromosome 14. The gross pathology in panlobular emphysema is widespread destruction of alveolar tissue with dilation of small airspaces throughout the lungs. Destruction is accentuated at the lung bases, with less severe destruction in the upper lobes. The histomorphology of centrilobular and panlobular emphysema is similar. “Free-floating” alveolar septa () are seen admixed with normal alveolar tissue in centrilobular emphysema. Normal alveolar tissue is not seen in panlobular.Citation7 With progressive destruction, the formation of bulla (airspace dilation in excess of 1 mm) is often observed. These two forms of emphysema are broadly classified as diffuse, and they can cause impairment of lung function.
Figure 2 Histologic features of centrilobular emphysema. A section of lung tissue shows fragmented and “free-floating” alveolar septa (arrow) characteristic of emphysema.
Other types of emphysema include localized (distal acinar) and paracicatricial (irregular) emphysema. These focal forms do not result in impairment of lung function but can cause spontaneous pneumothoraces. Grossly, localized emphysema has one or two sites of severe lung parenchyma destruction, most commonly located at the extreme apex of the lung. Free-floating alveolar septa and bulla are again the histomorphologic correlate to the gross pathology. Paracicatricial emphysema is defined as alveolar destruction surrounding foci of scarred lung tissue and can be seen in a wide variety of pulmonary disorders from healed foci of infection to the interstitial pneumonias as well as pneumoconioses.
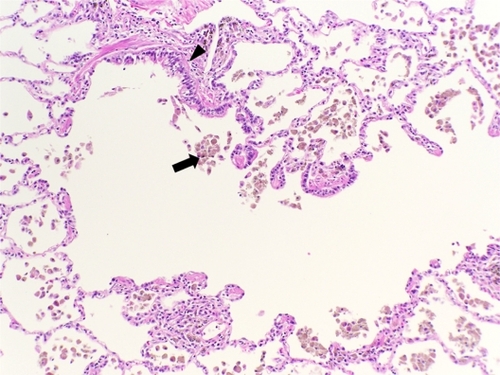
Small airway obstruction has also been recognized as an important pathologic finding associated with COPD ().Citation8,Citation9 Small airways disease includes respiratory bronchiolitis and membranous bronchiolitis.Citation10 The histomorphology includes collections of macrophages containing smoker’s pigment within respiratory bronchiole lumina, alveolar ducts, and alveoli. A lymphocytic infiltrate within bronchiolar walls and peribronchiolar fibrosis with bronchiolar metaplasia of alveolar septa (lambertosis) are additional histologic features. Goblet cell metaplasia and mucostasis may also be seen within the membranous bronchioles.
Figure 3 Histologic features of small airways disease. A section of lung tissue shows accumulation of macrophages with smoker’s pigment (arrow) within and around a respiratory bronchiole (arrow head).
Patients with COPD exhibit different characteristic features of chronic bronchitis, emphysema, and/or asthma, but all exhibit clinical symptoms of airflow obstruction. The Global Initiative for Chronic Obstructive Lung Disease (GOLD) has developed a COPD disease severity classification, stages I–IV, based primarily on pulmonary function tests (ie, forced vital capacity [FVC] and forced expiratory volume in 1 second [FEV1]). Pulmonary mechanics provide a clinical “snapshot” reflecting the combined effect of pathologic changes in all the affected lung compartments. Quantitative or high resolution computed tomography (CT) are surrogate measures for lung histology and are useful to assess airway wall thickening and emphysema.Citation11,Citation12 In the absence of actual tissue specimens, these two CT measurements provide an “ex vivo” type of pathological characterization. Similarly, CT assessment of airway wall thickness and emphysema each independently show associations with respiratory symptoms, such as dyspnea, cough, and wheezing, in COPD patients.Citation11
Cigarette smoking is the primary environmental risk factor for COPD. In addition, other environmental (eg, wood smoke) and occupational exposures as well as genetics contribute to COPD pathogenesis. Consequently, pathologic changes and clinical symptoms are linked to the interaction of host factors with the environment. These interactions generate the pathologic triad of COPD: persistent inflammation, protease–antiprotease imbalance, and oxidative stress. This triad results in mucous/goblet cell metaplasia and hyperplasia, mucous hypersecretion, fibrosis, smooth-muscle alterations, and lung-tissue destruction.Citation1 For this review, the authors have limited their discussion to a review of molecular markers and genetic host factors and their association with COPD histopathology and disease status. The Human Genome Project and the advancement of molecular biology and genetic analysis techniques, microarray analyses, and genome-wide association studies (GWASs) are leading to new discoveries of genetic variations that predispose to COPD susceptibility, lung function, and overall disease severity. Similar to the biomarkers, these genetic polymorphisms are linked to inflammation, protease–antiprotease imbalance, and oxidative stress.
Mechanistic triad: oxidative stress, protease–antiprotease imbalance, and inflammation
Oxidative stress
Chronic smoking exposes the respiratory tree and lungs to reactive oxygen species (ROS), resulting in oxidative stress and injury. This triggers production of other ROS and lipid peroxidation and subsequent pulmonary inflammation.Citation13 For example, increased expression of 4-hydroxy-2-nonenal, a product of lipid peroxidation, is present in both the airway and alveolar epithelia of COPD patients.Citation14 Similarly, malondialdehyde (MDA), an end product of lipid peroxidation also increases in the blood of COPD patients and increases with severity of disease.Citation15 Both tobacco and wood smoke exposure-associated COPD will trigger MDA production. Recent studies demonstrate that in smoke-associated COPD patients, increases in blood MDA inversely correlate with changes in FEV1.Citation16
Cigarette smoking also causes particulate deposits in the lungs with corresponding increases in tissue iron (by Perl’s Prussian blue staining).Citation17 Compared with nonsmokers and healthy smokers, smokers with COPD have increased iron and ferritin and decreased transferrin in their bronchoalveolar lavage (BAL) fluid.Citation17 The authors of this paper have recently reported similar findings in A1ATD patients.Citation18 Importantly, Ghio et al demonstrated that cigarette smoke triggers ROS production by an iron-dependent mechanism.Citation17 Smoking triggers airway and pulmonary inflammation, with an influx of inflammatory cells including macrophages and neutrophils. The authors of this paper and others have reported that neutrophil elastase (NE) will degrade endogenous iron storage or transport proteins, ferritin, transferrin (TF), and lactoferrin, and therefore, may increase the “free” iron burden. Investigators demonstrated the NE-induced cleavage of TF and lactoferrin in vitro.Citation19 The authors of this paper have recently reported that NE degrades ferritin to release “free” iron that can be taken up by airway epithelial cells.Citation18 NE degrades the ferritin very rapidly, and iron is taken up by the cells within a few hours. If there is redox-active iron in the airway surface liquid, the iron can mediate the Haber–Weiss reaction, converting any superoxide and/or hydrogen peroxide released during a neutrophilic respiratory burst to the highly cytotoxic hydroxyl radical.Citation20 Protease degradation of TF has been shown to result in iron-mediated hydroxyl radical formation and oxidative injury.Citation21,Citation22 Rodent models demonstrate that either cigarette smoke exposure or a single intratracheal installation of NE causes an increase in iron and ferritin in BAL and/or lungs of these animals, which can lead to ROS production and oxidative injury.Citation17,Citation18 In addition, genetic analyses with different cohorts of COPD subjects, both familial and nonfamilial cohorts as well as a smoker control group, demonstrated increased expression of iron regulatory protein 2 (IREB2 or IRP2) in the lungs of COPD patients compared with controls and a genetic association of IREB2 as a COPD susceptibility gene.Citation23 Collectively, this information highlights the importance of iron-mediated oxidative stress in COPD pathogenesis.
Smoking increases the oxidative burden to the lungs. In COPD, this is compounded by alterations in the antioxidant defenses. Data mining of genetic studies reveals alterations in the nuclear factor-erythroid 2-related factor (Nrf2)-mediated oxidative stress response pathway as well as the mitochondrial dysfunction pathway.Citation24 Nrf2 is a transcription factor that regulates Phase II and antioxidant genes in response to oxidant stress.Citation25,Citation26 In COPD subjects compared with healthy smokers, there is decreased expression of heme oxygenase 1 (HMOX-1) and catalase, components of the Nrf2 response pathway, and decreases in catalase and cytochrome C oxidase subunits, components of the mitochondrial dysfunction pathway. The HMOX-1 has a (GT)n dinucleotide repeat in the 5′-flanking region of the gene that has been reported to be highly polymorphic.Citation27 In a Japanese population study, a HMOX-1 promoter polymorphism with 30 or more GT dinucleotide repeats is associated with increased susceptibility to emphysema in smokers.Citation28 Furthermore, these investigators demonstrated that in response to oxidative stress, this polymorphism resulted in reduced transcriptional activity of the HMOX-1 gene decreasing the antioxidant capacity. Similarly, single-nucleotide polymorphisms (SNPs) in the glutathione-S-transferase (GST) P1 gene have been associated with apical distribution of emphysema.Citation29 Interestingly, increased iron and ferritin levels have also been reported in the upper lobes of smokers,Citation30 suggesting genetic variations along with iron-catalyzed oxidative stress/injury are important mechanisms in emphysema pathogenesis.
The association of emphysema with genetic variations in other antioxidant genes has also been reported. There are three forms of superoxide dismutase, SOD1 (Cu/Zn SOD), SOD2 (MnSOD) and SOD3 (extracellular SOD, ecSOD). SNPs in SOD3 have been reported to either protect or promote development of emphysema and COPD. In a study on populations of European descent, there was a higher frequency of the R213G (213Gly) variant of SOD3 in smokers who did not develop COPD, compared with individuals with COPD.Citation31 Similarly, in a Danish population study, smokers expressing the R213G polymorphism in the SOD3 gene have a reduced risk of COPD development.Citation32 However, other SOD3 polymorphisms are associated with increased risk of emphysema and reduced lung function. In two Copenhagen populations, two polymorphisms, E1 (rs8192287), in the 5′ untranslated region of exon 1, and I1 (rs9192288), in the first intron, are linked to decreased lung function.Citation33 In both populations, individuals homozygous for these polymorphisms, that is having both E1 and I1 polymorphisms have lower FVC% predicted. The investigators also followed these individuals for 26 years to monitor for COPD-associated hospitalizations and death. Individuals homozygous for these polymorphisms had an increased risk of COPD hospitalizations compared with noncarrier smokers. A group of US investigators with different populations reported related findings to the study in Copenhagen. These investigators looked at the same three reported polymorphisms, E1, I1, and R213G in severe COPD subjects, smoking controls, and two family-based cohorts.Citation34 Sorheim et al demonstrated that SOD3 SNPs E1 and I1 are associated with a higher incidence of CT-defined emphysema in one population, and only the E1 SNP is associated with more emphysema in a family-based population.Citation34 The same family-based cohort also demonstrates that E1 SNP is associated with increased airway wall thickness. The investigators concluded that the E1 and I1 SOD3 SNPs are associated with CT-documented emphysema, but not COPD susceptibility.Citation34
Markers of oxidant injury have also been linked to emphysema. One of these markers is the oxidation of guanine in deoxyribonucleic acid (DNA) or ribonucleic acid (RNA) to produce 8-oxo-7,8-dihydro-2′-deoxyguanosine (8OHdG) in DNA and 8-oxo-7,8-dihydroguanosine (8OHG) in RNA. Evaluation of emphysematous lungs from COPD patients with and without A1ATD reveals increased immunohistochemical staining for 8OHdG and 8OHG in the emphysematous lungs compared with non-COPD lungs.Citation35 In addition, lungs from A1ATD have increased oxidized DNA/RNA markers in alveolar macrophages and airway epithelial cells. These results suggest a role for nucleic acid oxidation in the pathophysiology of emphysema.
The free radical theory of aging suggests that reactive oxygen species, modifiable by genetic and environmental factors (eg, smoking, pollutants), mediate aging-associated alterations in cells and tissues.Citation36–Citation39 Senescence and apoptosis are also linked with aging and COPD.Citation40 Consequently, evidence of senescence markers and alveolar wall (ie, endothelial and/or epithelial) cell apoptosis in emphysematous lungs, implicates an aging paradigm important in COPD.Citation41
Cigarette smoking has been implicated as a major risk factor for pulmonary cell senescence. Exposing lung fibroblasts and human alveolar epithelial cells to tobacco smoke in vitro induces senescence, a complete loss of replicative capacity, in both cell types.Citation42,Citation43 In the alveolar epithelial cells, senescence is characterized by increased senescence-associated β-galactosidase activity, accumulation of lipofuscin, and increased expression of the cyclin-dependent kinase inhibitor, p21CIP1 (p21).Citation43 Similar findings are also seen in the lung fibroblasts, as well as upregulation of a different cyclin-dependent kinase inhibitor, p16INK4a (p16), a known senescence biomarker.Citation42 These reports were followed by studies using human lung emphysema tissues. Compared with healthy smokers and nonsmoker controls, emphysema patients have increased expression of senescence markers p16 and p21 in alveolar type II cells and alveolar endothelial cells.Citation44 These investigators, using fluorescent in situ hybridization to assess telomere length, also demonstrated telomere shortening, as a marker of senescence, in the alveolar type II cells and endothelial cells. Similarly, in COPD patients compared with healthy smoker and nonsmoker controls, telomere shortening occurs in circulating leukocytes.Citation45 A SNP in the BICD1 (bicaudal D homolog 1) gene is associated with human telomere length variations.Citation46 Other SNPs were evaluated in several large cohorts by GWAS, comparing mild, moderate, and severe emphysema patients with controls.Citation47 High-resolution CT (HRCT) was used to characterize the severity of the emphysema. This group also found a SNP in BICD1 that is significantly different between emphysema patients and controls and thus implicates this gene as an emphysema susceptibility gene.Citation47
In addition to increased senescence in emphysematous lungs, there is increased apoptosis as part of the pathophysiology of emphysema and COPD. In particular, there is apoptosis of the structural cells, alveolar epithelial and endothelial cells. Apoptosis compounded by loss of proliferative capacity in these diseased lungs results in lung tissue loss and destruction (reviewed in Demedts et alCitation48 and Morissette et alCitation49). Emphysematous lungs, compared with those from healthy smokers and nonsmokers, have increased terminal transferase dUTP nick end labeling staining in alveolar septa in both alveolar epithelial and endothelial cells.Citation50 The authors confirmed these findings with another immunohistochemical indicator of enhanced apoptosis, single-stranded DNA, demonstrating increased immunohistochemical staining in emphysematous lungs.Citation50 Importantly, these investigators confirmed earlier in vitro and animal studies demonstrating decreased expression of vascular endothelial growth factor (VEGF) and VEGF R2 receptor in the pathogenesis of endothelial apoptosis in the emphysematous lungs.Citation50 Other apoptosis markers are present in emphysematous lungs. There are increased p53 protein levels, Bax/Bcl-xL ratio and tumor necrosis factor-related apoptosis-inducing ligand receptors 1, 2, and 3 protein levels in emphysematous lungs.Citation51 Interestingly, different SNPs in the cell division cycle 6 homolog (CDC6) gene correspond to a gene-dosage effect in annual lung function decline in ex-smoking COPD patients.Citation52 For example, SNP6 patients with Val/Val have a 2% annual rate of decline in FEV1% predicted, while patients with Ile/Ile have an 11% annual rate of decline.
Another link to apoptosis in COPD is the autophagic process. Autophagy is a highly regulated process by which internal organelles and proteins are degraded. Autophagic proteins Beclin 1 and LC3B mediate cigarette smoke extract-induced apopotsis of airway epithelial cells.Citation53 In addition, by Western analysis, electron microscopy, and immunoflourescence, there is increased expression of the autophagic proteins LC3B-II/LC3B-I, Atg4, Atg5-artg12, and Atg7, as well as increased numbers of different stages of autophagic vacuoles in the lungs of COPD patients in all GOLD categories, with a corresponding increase in caspase 3 activity, a marker of apoptosis in GOLD 3/4 patients.Citation54
Inflammation
Inflammation in COPD is not a separate entity by itself but is integrally related to oxidative stress and protease–antiprotease imbalance. Cigarette smoking and other inhaled pollutants can serve as a trigger or activator of inflammation, oxidative stress, and protease–antiprotease imbalance.
COPD patients compared with control subjects experience an overall heightened state of systemic inflammation reflected by biomarkers, such as cytokines and nitric oxide, which further increase with exacerbations.Citation55 Similarly, bronchoalveolar lavage fluid and sputum also demonstrate increased inflammatory biomarkers such as cytokines, proteases, and soluble cytokine receptors.Citation56 In addition, cytokine levels can increase with COPD disease severity.Citation57
Cigarette smoking and perhaps other types of smoke inhalation trigger an inflammatory response in the lungs resulting in the influx of different types of inflammatory cells. Neutrophils, macrophages, T lymphocytes, as well as eosinophils and mast cells, have all been associated with chronic bronchitis or COPD.Citation58 It is interesting to note that there is a strong immunologic aspect to COPD that is characterized by T cell activation, proliferation into effector-type T cells, and then chemoattraction to the lung based on tissue-specific chemokine receptor and ligand expression. Furthermore, immune tolerance mechanisms, specifically failure to develop tolerance, correlates with COPD disease severity.Citation59 Bronchial biopsies or surgical resection specimens of airways and lungs have been evaluated by immunohistochemistry to identify the infiltrating cells in these tissues. Bronchial biopsies from chronic bronchitis subjects with airflow obstruction (COPD) compared with normal subjects demonstrate increased numbers of CD3+ and CD8+ T cells, macrophages, and increased HLA-DR expression.Citation60 Importantly, these investigators demonstrate an inverse relationship between the number of CD8+ T cells, neutrophils, and eosinophils with FEV1% predicted in the chronic bronchitis patients. Similarly, both airways and lungs from smokers with COPD, compared with normal subjects, have increased CD8+ T cells in the lung parenchyma, pulmonary arteries, and peripheral airways.Citation61,Citation62 Importantly, the number of CD8+ T cells in the lung parenchyma and pulmonary arteries of the COPD patients negatively correlates with FEV1% predicted.Citation61 Furthermore, compared with control subjects, COPD subjects have increased expression of CXCR3+ cells, important in a Th1-type of response, which are also CD8+, in the airway epithelium and submucosa and increased expression of the CXCR3-ligand CXCL10 on the bronchiolar epithelium.Citation63 These studies highlight the importance of T cells in progression of COPD.
Neutrophils are also an important infiltrating inflammatory cell in COPD. Bronchial biopsies from GOLD stage III and IV COPD patients (severe/very severe) compared with healthy nonsmokers and healthy smokers, demonstrate increased expression of the chemokine CCL5, leukocyte chemotactic chemokine, in the airway epithelium and a corresponding increase in neutrophils in the airway submucosa.Citation64 Interestingly, based on HRCT scoring of emphysema, an SNP in the CCL5 gene has been associated with milder emphysema.Citation65 The submucosal neutrophils also have increased expression of CD11b and CD44, which promote adhesiveness in severe/very severe COPD.Citation64 The severity of COPD is also aggravated by acute exacerbations, which are often associated with microbial infections and neutrophilic inflammation.Citation66 Bronchial biopsies from COPD patients with acute severe exacerbations were compared with biopsies from stable COPD patients as well as normal nonsmoker, nonatopic control subjects. Compared with stable COPD and control subjects, patients with acute severe exacerbations have neutrophilia and, by in situ hybridization, increased mRNA expression of CXCL5 (epithelial derived neutrophil attractant-78), CXCL8 (IL-8), CXCR1, and CXCR2.Citation67 In addition, a SNP in the CCL1 gene, a chemokine that is chemotactic for leukocytes, is significantly associated with the frequency of acute severe COPD exacerbations.Citation68 Furthermore, prospectively, these investigators demonstrated this polymorphism is significantly associated with an increased risk of death due to acute exacerbations. Consequently, they demonstrated a “gene–dosage effect” between subjects that are heterozygous versus homozygous for the polymorphism.Citation68 Altogether, these studies highlight the key role of inflammatory cells, particularly neutrophils, macrophages, and T cells, in mediating progression of COPD.
Cigarette smoking is a trigger for senescence. One of the sequelae of senescence is the development of a pro-inflammatory phenotype.Citation69 In lung tissues from COPD patients compared with healthy smoker and nonsmoking control subjects, nuclear factor (NF)-κB activation is increased in senescent alveolar type II cells.Citation70 In addition, compared with nonsmoking controls, there is decreased sirtuin deacetylase (SIRT1) protein expression, an anti-inflammatory and anti-aging protein that negatively regulates transcription factors such as NF-κB, in the lungs of COPD patients.Citation71 Thus, senescent cells may propagate the inflammatory response in COPD. Importantly, alveolar and endothelial senescence markers, p16 and p21, are positively correlated with airflow obstruction (FEV1% predicted)Citation44 and shortened telomere length in leukocytes from COPD patients with hypoxemia.Citation45
Protease–antiprotease imbalance
In a simplistic sense, emphysema is caused by the imbalance of proteases and antiproteases that results in lung parenchymal destruction.Citation72 The classic example is A1ATD, where unopposed neutrophil elastase has deleterious effects on the lungs. However, in the overall pathogenesis, it is the interactions between ROS and proteases or antiproteases that trigger this imbalance. Smoke exposure from cigarettes or biomass exposure from indoor fires can inactivate endogenous antiproteases,Citation73 as well as trigger an acute pulmonary response that activates resident alveolar macrophages and promotes neutrophil influx into lungs. As the smoke exposure continues and becomes more chronic, there is continued accumulation of macrophages, neutrophils, and CD8+ T cells in the lungs.Citation74 The macrophages and neutrophils release a variety of proteases, including neutrophil elastase, proteinase 3, matrix metalloproteinases (MMPs), and cathepsins. These proteinases “support” each other by activating each other or inhibiting their endogenous inhibitors, such as neutrophil elastase inhibiting tissue inhibitors of MMPs, and MMPs degrading α1-antitrypsin.Citation74 These proteinases cleave components of the extracellular matrix, elastin fibers and collagen, generating elastin fragments or collagen-derived peptides such as proline-glycine-proline, which have been shown to be chemotactic for monocytes, the precursor cell for macrophagesCitation75 or neutrophils.Citation76 Collectively, chemotactic peptide fragments perpetuate macrophage and neutrophil accumulation and lung destruction.
Genetic factors also regulate protease activity in the lung and emphysema development. MMP12 knockout mice are protected from cigarette-smoke-induced emphysema.Citation77 Genetic studies in large COPD-related cohorts, including a family-based COPD cohort, identified a SNP in MMP12 that protects lung function and reduces the risk of COPD in adult smokers.Citation78 In contrast, two studies from Japan that used CT or HRCT to characterize the presence and severity of emphysema identified an SNP in MMP9 that is associated with smoking-induced emphysema development.Citation79,Citation80 In addition to MMPs, SERPINE2, an inhibitor of MMP activation and extracellular matrix destruction, has been linked to COPD as a potential susceptibility gene.Citation81,Citation82 Both studies used large case–control cohorts as well as family-based cohorts to demonstrate the association of SERPINE2 SNPs with COPD. DeMeo et al proposed a gene-by-smoking interaction for this gene.Citation81
Summary
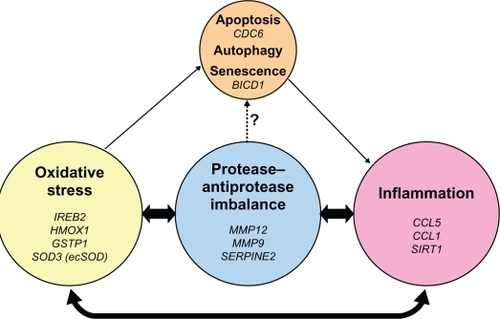
The unique histopathology of COPD is driven by interactions between host factors and environmental exposures. Basic and translational research have provided important discoveries concerning these interactions in COPD, resulting in the development of biomarkers reflecting protease injury, oxidative stress, inflammation, senescence, and apoptosis (). The breadth of pathological variability in COPD presents a challenge for clinicians. Unique gene and environmental interactions may provide critical new insights explaining individual manifestations of disease and opportunities to improve outcomes for patients.
Figure 4 Pathogenic triad of COPD: oxidative stress, protease–antiprotease imbalance, and inflammation. Oxidative stress, protease–antiprotease imbalance and inflammation each are important in the pathogenesis of COPD; however, they constantly interact and may at times overlap with each other in the overall pathogenesis of COPD. As a consequence of oxidative stress, in particular cigarette smoking-induced oxidative stress, apoptosis, autophagy, and senescence are each potential lung cell fates. Senescent cells express a pro-inflammatory phenotype. Proteases, such as neutrophil elastase, have been shown in vitro to induce airway epithelial apoptosis,Citation83 but this relationship has not yet been specifically demonstrated in human subjects. Listed in italics are the genetic polymorphisms that have been reported and discussed in this review, to be associated with COPD or emphysema in that area of the pathogenic triad.
Acknowledgements
This work was supported by National Institutes of Health grant ES016836, the Alpha-1 Foundation, and the Duke School of Medicine.
Disclosure
The authors report no conflicts of interest in this work.
References
- WrightJLChurgAPathologic features of chronic obstructive pulmonary disease: diagnostic criteria and differential diagnosisFishmanAPEliasJAFishmanJAGrippiMASeniorRMPackAIFishman’s pulmonary diseases and disorders14th edNew YorkMcGraw Hill2008693705
- RoggliVCaglePEmphysema and chronic bronchitisTomashefskiJCaglePFarverCFraireADail and hammar’s pulmonary pathology3rd edNew YorkSpringer2008866885
- American Thoracic SocietyStandards for the diagnosis and care of patients with chronic obstructive pulmonary diseaseAm J Respir Crit Care Med19951525 Pt 2S77S1217582322
- ReidLMeasurement of the bronchial mucous gland layer: a diagnostic yardstick in chronic bronchitisThorax19601513214114437095
- PrattPEmphysema and chronic airways diseaseDailDHammarSPulmonary pathology2nd edNew YorkSpringer-Verlag1994847865
- ThurlbeckWChronic airflow obstructionThurlbeckWChurgAPathology of the lung2nd edNew YorkThieme Medical Publishers1995739825
- GreenLEmphysema and diseases of large airwaysZanderDFarverCPulmonary pathologyPhiladelphia, PAChurchill Livingstone/Elsevier2008416433
- HoggJCChuFUtokaparchSThe nature of small-airway obstruction in chronic obstructive pulmonary diseaseN Engl J Med20043502645265315215480
- HoggJCTimensWThe pathology of chronic obstructive pulmonary diseaseAnnu Rev Pathol2009443545918954287
- CaglePRoggliVPathology of small airwaysTomashefskiJFarverCFraireACaglePDail and Hammar’s pulmonary pathology13rd edNew YorkSpringer2008886910
- GrydelandTBDirksenACoxsonHOQuantitative computed tomography measures of emphysema and airway wall thickness are related to respiratory symptomsAm J Respir Crit Care Med201018135335919926869
- PatelBDCoxsonHOPillaiSGAirway wall thickening and emphysema show independent familial aggregation in chronic obstructive pulmonary diseaseAm J Respir Crit Care Med200817850050518565956
- RahmanIAdcockIMOxidative stress and redox regulation of lung inflammation in COPDEur Respir J20062821924216816350
- RahmanIvan SchadewijkAACrowtherAJ4-Hydroxy-2-nonenal, a specific lipid peroxidation product, is elevated in lungs of patients with chronic obstructive pulmonary diseaseAm J Respir Crit Care Med200216649049512186826
- KluchovaZPetrasovaDJoppaPDorkovaZTkacovaRThe association between oxidative stress and obstructive lung impairment in patients with COPDPhysiol Res200756515616497100
- MontanoMCisnerosJRamirez-VenegasAMalondialdehyde and superoxide dismutase correlate with FEV(1) in patients with COPD associated with wood smoke exposure and tobacco smokingInhal Toxicol20102286887420583895
- GhioAJHilbornEDStonehuernerJGParticulate matter in cigarette smoke alters iron homeostasis to produce a biological effectAm J Respir Crit Care Med20081781130113818723436
- FischerBMDomowiczDAZhengSNeutrophil elastase increases airway epithelial nonheme iron levelsClin Transl Sci2009233333920411049
- BritiganBEHayekMBDoebbelingBNFickRBJrTransferrin and lactoferrin undergo proteolytic cleavage in the Pseudomonas aeruginosa-infected lungs of patients with cystic fibrosisInfect Immun199361504950558225581
- OwenCAProteinases and oxidants as targets in the treatment of chronic obstructive pulmonary diseaseProc Am Thorac Soc20052373385 discussion 394–37516267366
- BritiganBEEdekerBLPseudomonas and neutrophil products modify transferrin and lactoferrin to create conditions that favor hydroxyl radical formationJ Clin Invest199188109211021655825
- MillerRABritiganBEProtease-cleaved iron-transferrin augments oxidant-mediated endothelial cell injury via hydroxyl radical formationJ Clin Invest199595249125007769095
- DeMeoDLMarianiTBhattacharyaSIntegration of genomic and genetic approaches implicates IREB2 as a COPD susceptibility geneAm J Hum Genet20098549350219800047
- ComandiniAMarzanoVCurradiGMarkers of anti-oxidant response in tobacco smoke exposed subjects: a data-mining reviewPulm Pharmacol Ther20102348249220594977
- KenslerTWWakabayashiNBiswalSCell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathwayAnnu Rev Pharmacol Toxicol2007478911616968214
- MalhotraDThimmulappaRNavas-AcienADecline in NRF2-regulated antioxidants in chronic obstructive pulmonary disease lungs due to loss of its positive regulator, DJ-1Am J Respir Crit Care Med200817859260418556627
- KimparaTTakedaAWatanabeKMicrosatellite polymorphism in the human heme oxygenase-1 gene promoter and its application in association studies with Alzheimer and Parkinson diseaseHum Genet19971001451479225984
- YamadaNYamayaMOkinagaSMicrosatellite polymorphism in the heme oxygenase-1 gene promoter is associated with susceptibility to emphysemaAm J Hum Genet20006618719510631150
- DeMeoDLHershCPHoffmanEAGenetic determinants of emphysema distribution in the national emphysema treatment trialAm J Respir Crit Care Med2007176424817363767
- NelsonMEO’Brien-LadnerARWesseliusLJRegional variation in iron and iron-binding proteins within the lungs of smokersAm J Respire Crit Care Med19961534 Pt 113531358
- YoungRPHopkinsRBlackPNFunctional variants of antioxidant genes in smokers with COPD and in those with normal lung functionThorax20066139439916467073
- JuulKTybjaerg-HansenAMarklundSLangePNordestgaardBGGenetically increased antioxidative protection and decreased chronic obstructive pulmonary diseaseAm J Respir Crit Care Med200617385886416399992
- DahlMBowlerRPJuulKSuperoxide dismutase 3 polymorphism associated with reduced lung function in two large populationsAm J Respir Crit Care Med200817890691218703790
- SorheimICDeMeoDLWashkoGPolymorphisms in the superoxide dismutase-3 gene are associated with emphysema in COPDCOPD2010726226820673035
- DesleeGWoodsJCMooreCOxidative damage to nucleic acids in severe emphysemaChest200913596597419118262
- HarmanDAging: a theory based on free radical and radiation chemistryJ Gerontol19561129830013332224
- HarmanDThe free radical theory of agingAntioxid Redox Signal2003555756114580310
- FinkelTHolbrookNJOxidants, oxidative stress and the biology of ageingNature200040823924711089981
- ItoKBarnesPJCOPD as a disease of accelerated lung agingChest200913517318019136405
- AoshibaKNagaiASenescence hypothesis for the pathogenetic mechanism of chronic obstructive pulmonary diseaseProc Am Thorac Soc2009659660119934355
- MacNeeWTuderRMNew paradigms in the pathogenesis of chronic obstructive pulmonary disease IProc Am Thorac Soc2009652753119741262
- NyunoyaTMonickMMKlingelhutzACigarette smoke induces cellular senescenceAm J Respir Cell Mol Biol20063568168816840774
- TsujiTAoshibaKNagaiACigarette smoke induces senescence in alveolar epithelial cellsAm J Respir Cell Mol Biol20043164364915333326
- TsujiTAoshibaKNagaiAAlveolar cell senescence in patients with pulmonary emphysemaAm J Respir Crit Care Med200617488689316888288
- SavaleLChaouatABastuji-GarinSShortened telomeres in circulating leukocytes of patients with chronic obstructive pulmonary diseaseAm J Respir Crit Care Med200917956657119179485
- ManginoMBrouiletteSBraundPA regulatory SNP of the BICD1 gene contributes to telomere length variation in humansHum Mol Genet2008172518252318487243
- KongXChoMHAndersonWGenome-wide association study identifies BICD1 as a susceptibility gene for emphysemaAm J Respir Crit Care Med2011183434920709820
- DemedtsIKDemoorTBrackeKRJoosGFBrusselleGGRole of apoptosis in the pathogenesis of COPD and pulmonary emphysemaRespir Res200675316571143
- MorissetteMCParentJMilotJAlveolar epithelial and endothelial cell apoptosis in emphysema: what we know and what we need to knowInt J Chron Obstruct Pulmon Dis20094193119436685
- KasaharaYTuderRMCoolCDEndothelial cell death and decreased expression of vascular endothelial growth factor and vascular endothelial growth factor receptor 2 in emphysemaAm J Respir Crit Care Med20011633 Pt 173774411254533
- MorissetteMCVachon-BeaudoinGParentJChakirJMilotJIncreased p53 level, Bax/Bcl-x(L) ratio, and TRAIL receptor expression in human emphysemaAm J Respir Crit Care Med200817824024718511705
- TakabatakeNToriyamaSIgarashiAA novel polymorphism in CDC6 is associated with the decline in lung function of ex-smokers in COPDBiochem Biophys Res Commun200938155455919233139
- KimHPWangXChenZHAutophagic proteins regulate cigarette smoke-induced apoptosis: protective role of heme oxygenase-1Autophagy2008488789518769149
- ChenZHKimHPSciurbaFCEgr-1 regulates autophagy in cigarette smoke-induced chronic obstructive pulmonary diseasePLoS One20083e331618830406
- KaradagFKarulABCildagOYilmazMOzcanHBiomarkers of systemic inflammation in stable and exacerbation phases of COPDLung200818640340918807087
- BarnesPJChowdhuryBKharitonovSAPulmonary biomarkers in chronic obstructive pulmonary diseaseAm J Respir Crit Care Med200617461416556692
- HacievliyagilSSGunenHMutluLCKarabulutABTemelIAssociation between cytokines in induced sputum and severity of chronic obstructive pulmonary diseaseRespir Med200610084685416214322
- O’DonnellRBreenDWilsonSDjukanovicRInflammatory cells in the airways in COPDThorax20066144845416648353
- CosioMGSaettaMAgustiAImmunologic aspects of chronic obstructive pulmonary diseaseN Engl J Med20093602445245419494220
- O’ShaughnessyTCAnsariTWBarnesNCJefferyPKInflammation in bronchial biopsies of subjects with chronic bronchitis: inverse relationship of CD8+ T lymphocytes with FEV1Am J Respir Crit Care Med19971558528579117016
- SaettaMBaraldoSCorbinoLCD8+ve cells in the lungs of smokers with chronic obstructive pulmonary diseaseAm J Respir Crit Care Med199916071171710430750
- SaettaMDi StefanoATuratoGCD8+ T-lymphocytes in peripheral airways of smokers with chronic obstructive pulmonary diseaseAm J Respir Crit Care Med19981573 Pt 18228269517597
- SaettaMMarianiMPanina-BordignonPIncreased expression of the chemokine receptor CXCR3 and its ligand CXCL10 in peripheral airways of smokers with chronic obstructive pulmonary diseaseAm J Respir Crit Care Med20021651404140912016104
- Di StefanoACaramoriGGnemmiIAssociation of increased CCL5 and CXCL7 chemokine expression with neutrophil activation in severe stable COPDThorax20096496897519703829
- HizawaNMakitaHNasuharaYFunctional single nucleotide polymorphisms of the CCL5 gene and nonemphysematous phenotype in COPD patientsEur Respir J20083237237818385174
- SethiSMalliaPJohnstonSLNew paradigms in the pathogenesis of chronic obstructive pulmonary disease IIProc Am Thorac Soc2009653253419741263
- QiuYZhuJBandiVBiopsy neutrophilia, neutrophil chemokine and receptor gene expression in severe exacerbations of chronic obstructive pulmonary diseaseAm J Respir Crit Care Med200316896897512857718
- TakabatakeNShibataYAbeSA single nucleotide polymorphism in the CCL1 gene predicts acute exacerbations in chronic obstructive pulmonary diseaseAm J Respir Crit Care Med200617487588516864713
- CampisiJd’Adda di FagagnaFCellular senescence: when bad things happen to good cellsNat Rev Mol Cell Biol2007872974017667954
- TsujiTAoshibaKNagaiAAlveolar cell senescence exacerbates pulmonary inflammation in patients with chronic obstructive pulmonary diseaseRespiration201080597020016134
- RajendrasozhanSYangSRKinnulaVLRahmanISIRT1, an antiinflammatory and antiaging protein, is decreased in lungs of patients with chronic obstructive pulmonary diseaseAm J Respir Crit Care Med200817786187018174544
- ChurgAWrightJLProteases and emphysemaCurr Opin Pulm Med20051115315915699789
- CavarraELucattelliMGambelliFHuman SLPI inactivation after cigarette smoke exposure in a new in vivo model of pulmonary oxidative stressAm J Physiol Lung Cell Mol Physiol2001281L412L41711435216
- ShapiroSDProteinases in chronic obstructive pulmonary diseaseBiochem Soc Trans2002309810212023833
- HunninghakeGWDavidsonJMRennardSElastin fragments attract macrophage precursors to diseased sites in pulmonary emphysemaScience19812129259277233186
- WeathingtonNMvan HouwelingenAHNoeragerBDA novel peptide CXCR ligand derived from extracellular matrix degradation during airway inflammationNat Med20061231732316474398
- HautamakiRDKobayashiDKSeniorRMShapiroSDRequirement for macrophage elastase for cigarette smoke-induced emphysema in miceScience1997277200220049302297
- HunninghakeGMChoMHTesfaigziYMMP12, lung function, and COPD in high-risk populationsN Engl J Med20093612599260820018959
- ItoINagaiSHandaTMatrix metalloproteinase-9 promoter polymorphism associated with upper lung dominant emphysemaAm J Respir Crit Care Med20051721378138216126934
- MinematsuNNakamuraHTatenoHNakajimaTYamaguchiKGenetic polymorphism in matrix metalloproteinase-9 and pulmonary emphysemaBiochem Biophys Res Commun200128911611911708786
- DemeoDLMarianiTJLangeCThe SERPINE2 gene is associated with chronic obstructive pulmonary diseaseAm J Hum Genet20067825326416358219
- ZhuGWarrenLAponteJThe SERPINE2 gene is associated with chronic obstructive pulmonary disease in two large populationsAm J Respir Crit Care Med200717616717317446335
- SuzukiTMoraesTJVachonEProteinase-activated receptor-1 mediates elastase-induced apoptosis of human lung epithelial cellsAm J Respir Cell Mol Biol20053323124715891109