
Abstract
The high incidence of chronic obstructive pulmonary disease (COPD), one of the most prevalent diseases worldwide, has attracted growing attention. Cigarette smoking is considered a major contributory factor in the pathogenesis and progression of COPD due to the tremendous oxidative burden that it causes, which induces an oxidant/antioxidant imbalance. Excessive oxidation induced by the excessive generation of mitochondrial reactive oxygen species disturbs the antioxidant systems and plays an important role in triggering and promoting chronic inflammation of airways. Given that mitochondria is one of the main sites of reactive oxygen species production by the oxidative phosphorylation process, oxidative stress may affect mitochondrial function by changing its structure and morphology and by affecting a series of mitochondrial proteins. In particular, PTEN-induced putative kinase 1/Parkin and p62 play critical roles in mitophagy. During the process, the Akt ubiquitin E3 ligase is an important mediator associated with cigarette smoke exposure-induced pulmonary endothelial cell death and dysfunction. Thus, understanding the underlying mechanisms of the signaling pathway may provide important information regarding the therapeutic treatment of COPD by application of alternative PTEN-induced putative kinase 1 targets or ubiquitin E3 ligase.
Introduction
Chronic obstructive pulmonary disease (COPD), an inflammatory disease with rising incidence worldwide, is characterized by sustained airflow limitation and is always associated with pulmonary arterial hypertension, hypoxemia, and hypercapnia, which significantly increases the likelihood of cardiopulmonary complications and aggravates the patients’ conditions. A survey showed that about 65 million people have COPD globally, and almost 3 million people, majorly from low- and middle-income countries, died of complications associated with COPD in 2005.Citation1
With regard to the causes of the development of COPD, various studies have proven that oxidative stress induced by reactive oxygen species (ROS) plays a major role in triggering and promoting chronic inflammation of airways and imbalances in protease/antiprotease activity.Citation2 Cigarette smoking is one of the most important risk factors that contain a large amount of ROS that induce an oxidant burden in smokers,Citation3–Citation7 affecting the oxidant–antioxidant mechanisms in the development of COPD.Citation8 Exposure to cigarette smoke induces the production of endogenous ROS as a byproduct of functional electron transfer chain in the mitochondria.Citation9 Mitochondria is a crucial semiautonomous cellular organelle housing the metabolic processes of oxidative phosphorylation and fatty acid metabolism, and is also a tool bridging signal transduction between the nucleus and extracellular threats. Despite the fact that mitochondria is one of the main sources of endogenous ROS, mitochondrial dysfunction has been found in oxidant-induced lung damage due to the susceptibility of mitochondrial DNA to oxidative damage, which consists of a reduction in mitochondrial biogenesis.Citation10,Citation11 Peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α), a central integrator of multiple signaling pathways, has been demonstrated to play a key role in the regulation of mitochondrial biogenesis and oxidative stress by a mitochondrial transcription factor A (TFAM).Citation12 These signaling factors regulating mitochondrial biogenesis are increased in COPD muscle, leading to a low muscle oxidative capacity in COPD.Citation10,Citation13
Oxidative stress affects not only mitochondrial biogenesis, but also the mitochondrial quality control in the pathogenesis of lung diseases.Citation14 A previous study indicated that ROS induced by cigarette smoke were involved in the alterations in mitochondrial network morphology.Citation15 Additionally, in the airway smooth muscle (ASM) from patients with an airway disease, mitochondria exhibited substantial morphologic defects and the balance between mitochondrial fission and fusion was disturbed. Similar alterations have also been observed in the mitochondria in airway epithelial cells of COPD patients exposed to chronic cigarette smoke.Citation16 Overall, mitochondria seem to play an important role in different aspects of cellular structure and function, as well as in ROS induced by alterations of mitochondrial morphology. However, the molecular mechanisms between the mitochondria-induced ROS and the pathologic process of COPD have not been fully understood. Thus, this review aims to discuss how oxidative stress affects the progression and pathogenesis of COPD, and the corresponding alterations in mitochondria-mediated structures in response to oxidative stress have also been reviewed.
Source of ROS
Oxidation is a chemical process in which electrons are removed from molecules; during the process, the leaked electrons contribute to generation of some highly reactive free radicals. Generally speaking, the accumulated ROS are derived in two ways: exogenous and endogenous ROS (). Exogenous ROS primarily originate from inhaled toxic gases or environmental pollutants such as car exhaust fumes, occupational exposure to dusts and cigarette smoking. The sources of endogenous ROS include mitochondrial respiration, peroxisomes, the NADPH oxidase (NOX) system and inflammatory cells,Citation17 while the oxidative phosphorylation process in the mitochondria is one of the major sources of endogenous ROS.Citation9,Citation18
Figure 1 Sources of ROS in vivo.
Abbreviations: COPD, chronic obstructive pulmonary disease; ROS, reactive oxidative specis; SOD, superoxide dismutase; Eos, eosnophils; MPNs, polymorphonuclear cells; AMs, alveolar macrophages.
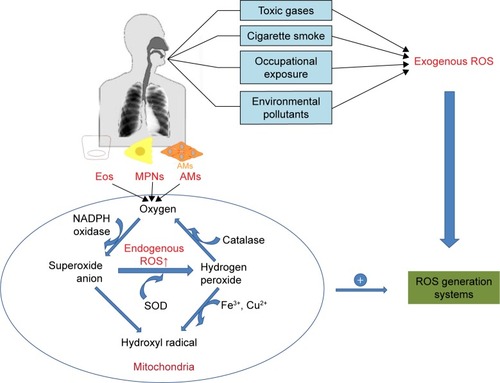
Peroxisome is a source of endogenous ROS, especially hydrogen peroxide (H2O2) and superoxide anions (O2−). For example, a heme-containing peroxidase in neutrophils and monocytes, myeloperoxidase, correlates with an increased level of ROS and chronic inflammation in COPD patients.Citation19,Citation20 Inflammatory cells also contribute to the formation of endogenous ROS under the catalysis of intracellular enzymes such as NOXs, ubiquinone reductase and cytochrome oxidase, while NOXs have long been recognized to be one of the primary generators of ROS within the lung and play pivotal roles in the pathogenesis of a number of diverse chronic lung disorders.Citation21,Citation22 NOX enzymes catalyze the conversion of O2 into O2−, which is a reactive radical characterized by its speedy interaction with intracellular macromolecules (lipids, proteins and DNA). There are seven members of NOXs: NOX1–5, dual oxidase DUOX1 and DUOX2. The expression levels of DUOX1 and DUOX2 that are primary expressed in the barrier epithelia were found to be low in the airway epithelium of former smokers with mild/moderate COPD.Citation23,Citation24 NOX1 mRNA expression was increased in patients with COPD, which contributes to rhinovirus infection-related exacerbations.Citation25 Pulmonary vascular endothelial cells have been shown to produce ROS in the presence of NOX2.Citation26 NOX3 in the lung was found to be associated with lung destruction and emphysema in aged mice with TLR4 deletion, but these effects were reversed on treatment with NOX inhibitors.Citation27 The ability of NOX4 to generate ROS has also been verified. An increasing number of researches have revealed that an increased expression of NOX4 was observed in pulmonary diseases.Citation28–Citation31 Recently, Hollins et al found that the expression of NOX4 was increased in ASM in COPD both in vivo and in primary cells in vitro.Citation32 Liu et al also showed NOX4 protein was increased in the ASM, and its expression correlated positively with disease severity and inversely correlated with the pulmonary functions in COPD patients.Citation33 Inhibition of NOX4 expression increased the mitochondrial TFAM level, leading to stimulation of mitochondrial biogenesis.Citation12 Oxidative stress induced by abundance of ROS can affect the organelles and disrupt cellular homeostasis.
Mitochondria are one of the main sources of production of ROS in eukaryotic cells. The production of ROS is a consequence of electron leakage occurring in the mitochondrial electron transport chain. During aerobic respiration, high-energy electrons pass through electron carriers in the respiratory chain complexes and react with molecular oxygen to form O2−, primarily at complexes I and III. This process is responsible for the generation of adenosine triphosphate (ATP), which promotes pumping of protons from the mitochondrial matrix into the intermembrane space, creating a measurable electrochemical proton gradient. However, protons are pumped back into the matrix in the presence of ATP synthase and react with the O2− in a reaction catalyzed by superoxide dismutase, contributing to the formation of hydrogen peroxide.Citation34 Superoxide at a high concentration causes oxidative damage. The formed H2O2 may give rise to hydroxyl radicals or is decomposed into water and oxygen by catalase. Therefore, the presence of O2− plays an important role in the formation of other forms of ROS. Excessive levels of mitochondrial ROS degrade the mitochondrial function and lead to the collapse of electrochemical gradient, which leaves the cell more susceptible to ROS-mediated injury.Citation35 Given that oxidative stress in COPD patients is a prevailing phenomenon, we next reviewed how oxidative stress induced by excessive ROS plays a role in the pathogenesis of inflammation in COPD.
The role of oxidative stress in COPD pathogenesis
Minutoli et alCitation36 and Park et alCitation37 suggested that accumulation of mitochondrial ROS activates inflammatory pathways in various tissues by interfering with mitochondrial quality control. Evidence shows that excessive mitochondrial ROS causes oxidative damage, which was reflected by the increased levels of damaged proteins, DNA and lipids in the mitochondria, as well as inflammatory markers within the cells in COPD patients.Citation38 Actually, ROS exhibit their toxic properties under specific conditions when the antioxidant system fails to eliminate excessive free radicals. In this case, gaseous-phase ROS in inhaled irritants cause direct damages to airway epithelial cells and their plasma cell membranes when its level exceeds the scavenge ability of the cellular anti-oxidative system.Citation39 Thus, antioxidant defense systems, including the nuclear factor-erythroid-2-related factor 2 (Nrf2)-mediated antioxidant system () and the phase II detoxifying enzymes, are attenuated by excessive accumulation of ROS. Denuded epithelia are always observed in COPD, increasing the possibility that the ROS can reach the ASM and promote the proliferation of ASM (), leading to thickening of the airway walls and an accelerated decline in lung function.Citation40
Figure 2 Antioxidant mechanisms of stress sensing in Nrf2-mediated system.
Abbreviation: Nrf2, nuclear factor-erythroid-2-related factor 2.
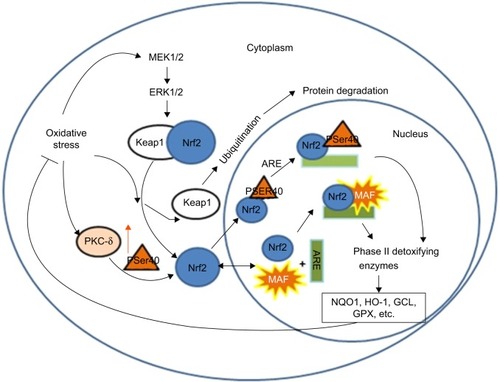
Figure 3 Mechanisms of oxidative stress injuries in COPD.
Abbreviations: ASM, airway smooth muscle; COPD, chronic obstructive pulmonary disease; IL, interleukin; MMP, matrix metalloproteinase; MUC5AC, mucin 5AC; NE, neutrophil elastase; ROS, reactive oxygen species; TNF, tumor necrosis factor.
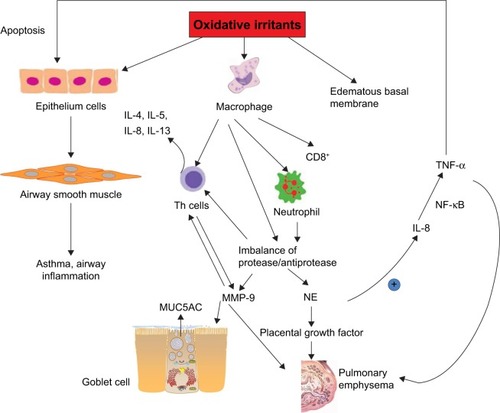
Inflammatory cells such as CD8+ T lymphocytes-neutrophils and alveolar macrophages are stimulated on exposure to oxidative stress, which affects the pulmonary epithelial cells.Citation41 One of the most obvious alterations in the inflammatory process is the imbalance of the protease/antiprotease system within the respiratory system, which promotes the release of the proteolytic enzymes of neutrophils and macrophages (matrix metalloproteinase-12), thus contributing to the degradation of their natural inhibitors. Consequently, the pulmonary parenchyma is destroyed, which accounts for the emphysema. During this process, accumulation of neutrophil elastase, a potent serine proteinase and a key effector that is most likely responsible for the emphysema, has been found in COPD patients.Citation42 Moreover, neutrophil elastase upregulates the levels of interleukin (IL)-8 and mucin 5AC that is a glycoprotein linked to mucus hypersecretion in the pulmonary tract. In bronchial epithelial and alveolar cells, ROS also induce the gene expression of inflammatory mediators such as tumor necrosis factor (TNF)-α, which in turn activates the transcription of IL-8, matrix metalloproteinase-9, mucin 5AC as well as interferon-γ.Citation43–Citation46 Several mechanisms such as TNFRs, NLRP3 inflammasome signals and MAPK are responsible for the mitochondrial ROS-mediated inflammatory responses. Bulua et al stated that mitochondrial ROS promote the production of proinflammatory cytokines including IL-6 and TNF in the cells from patients with TNFR1-associated periodic syndrome.Citation47 Levels of IL-1β and IL-18 are increased by the NLRP3 pathway. However, redox-sensitive transcription factors including NF-κB and AP-1 regulate these inflammatory genes’ expression.
Alterations of mitochondrial morphology in COPD
Recently, alterations in mitochondrial morphology are thought to be involved in oxidative stress in COPD pathology. The structural changes of mitochondria are closely linked to oxidative stress-mediated signaling. Cigarette smoke induced mitochondrial fragmentation and damaged their networked morphology in nonasthmatic human ASM cells, which depended on the level of oxidative stress.Citation16 However, cigarette smoke-triggered oxidative stress at a mild level induced mitochondrial hyperfusion in alveolar epithelial cells, which renders the cell more vulnerable to additional stress.Citation48 Furthermore, cigarette smoke-induced mitochondrial hyperfusion diminished mitochondrial quality control and impaired cellular stress resistance and cellular senescence, contributing to age-related COPD pathogenesis.
Thus, it is necessary to understand how the mitochondrial morphology is altered in response to the cellular stress induced by ROS in the progression of COPD.
To provide protection against severe cellular stress, mammalian mitochondria are not static structures, but undergo frequent shape changes to form tubular, reticular or networked morphology by changing mitochondrial fission/fusion. Both mitochondrial hyperfusion and mitochondrial fragmentation are a part of the mitochondrial quality control to maintain robust mitochondrial function for impaired mitochondria, while mitochondrial fusion is an adaptive stress-resolving mechanism that involves exchanging the damaged mitochondrial DNA, lipids or proteins with those of healthy mitochondria.Citation49
Mitochondrial fusion leads to elongated mitochondria in order to eliminate defective mitochondrial components.Citation50 During this process (), three large guanosine triphosphatases (GTPases), including mitofusion (Mfn) 1 and 2 embedded in the outer membrane of mitochondria and fusion protein (optic atrophy protein 1 [Opa1]) on the inner membrane of mitochondria, are of particular interest in morphologic maintenance. In fact, Opa1 and Mnfs share a reciprocal relationship. The evolutionarily conserved GTPases, Mfn1 and Mfn2, are responsible for outer mitochondrial membrane fusion,Citation51 while the pro-fusion effect of Mfn1 is promoted by the binding to guanine nucleotides to protein-β subunit 2 (Gβ2), thus facilitating and stabilizing clustering of Mfn1 on the outer mitochondrial membrane.Citation52 Furthermore, the effects of Mfn2 are attributed to the expression of PGC1-α and PGC1-β.Citation53 The dynamin-related GTPase, Opa1, mediates inner membrane fusion, and depletion of this protein results in severely reduced respiratory efficiency.Citation54 The effects of these three proteins have been proven by mouse knockout studies, which provided clear evidence of reduced mitochondrial fusion as an adaptive mechanism of resolving cellular stress in the cells.Citation55–Citation57 Hoffmann et alCitation16 suggested that cigarette smoke induced morphologic changes in the COPD epithelium by significantly increasing the expression of Mfn1/2 and Opa1, which were accompanied by increased levels of the proinflammatory mediators such as IL-8, IL-6 and IL-1β. Such a phenomenon of mitochondrial hyperfusion was also observed in the alveolar epithelial cells on exposure of cigarette smoke, which renders the cells more vulnerable to additional stress.Citation48 Furthermore, cigarette smoke-induced mitochondrial hyperfusion diminished mitochondrial quality control and impaired cellular stress resistance and cellular senescence, contributing to age-related COPD pathogenesis. Ahmad et al have shown that cigarette smoke extract induced elongated mitochondria and reduced ATP levels in lung epithelial cells and fibroblasts.Citation58 Mitochondrial fusion is a common phenomenon in mitochondria exposed to mild oxidative stress, whereas mitochondrial fission accelerates cellular necroptosis.Citation15
Figure 4 Schematic diagram shows that cigarette smoke induces mitochondrial fusion/fission and mitophagy.
Abbreviations: Mfn, mitofusion; Opa1, optic atrophy protein 1; PINK1, PTEN-induced putative kinase 1; ROS, reactive oxygen species; Ub, ubiquitin.
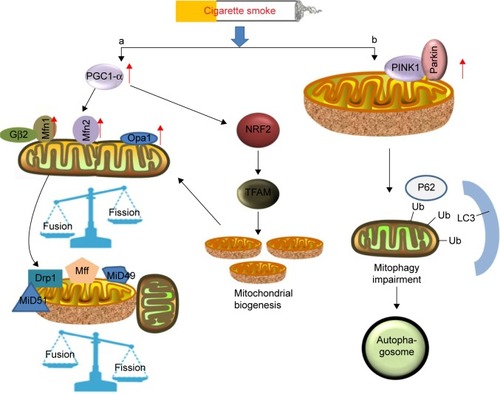
Accordingly, mitochondrial fission is highly correlated with cell apoptosis and mitophagy. Mitochondrial fission is achieved by phosphorylation of Drp1 at Ser616, which promotes the recruitment of Drp1 from the cytosol to the mitochondrial surface, by human fission protein-1, mitochondrial fission factor and mitochondrial dynamics proteins of 49 and 51.Citation14,Citation54 The possible mechanism indicates that oxidative stress increases the expression of the TFAM protein, thus triggering mitochondrial fission and loss by enhancing Drp1 translocation from the cytosol. Cigarette smoking-induced mitochondrial ROS accelerates the course of phosphorylation of Drp1.Citation15 However, inhibition of Drp1 significantly increases Bad S112 phosphorylation, which acts against fission-mediated apoptotic cell death.Citation59 Additionally, a prolonged exposure of cigarette smoke in COPD patients also significantly induces the expression of Fis1 and decreases the levels of Mfn1/2 and Opa1 on the airway epithelial mitochondria,Citation59 indicating that ROS contribute to mitochondrial fission. Mitochondrial fragmentation has further been described in the ASM cells of patients with COPD.Citation60 ASM cells cultured from a restricted group of COPD patients showed a collapsed mitochondrial membrane potential, an increased expression of mitochondrial fission protein and reduced respiratory complex protein, and accumulation of mitochondrial ROS, which contribute to the development of airway hyperresponsiveness and lung inflammation.Citation60 Moreover, Aravamudan et al also demonstrated that cigarette smoke induced mitochondrial fragmentation and damaged their networked morphology by increased expression of Drp1 and decreased Mfn2 in nonasthmatic human ASM cells, and this process involved ROS.Citation15 Recently, Wang et alCitation61 demonstrated that stress-induced mitochondrial fission and apoptosis were mediated by cyclin C-Cdk8 by enhancing the association between Drp1 and mitochondrial fission factor, which stabilizes their retention on the mitochondria in mouse embryonic fibroblasts. However, whether the same effects of C-Cdk8 are linked to lung disease remains unknown and deserves further investigation.
Mitophagy in COPD
Oxidative stress impairs mitochondrial function. When the depolarized or fragmented mitochondria are unable to fuse back into the functional pool of mitochondria, mitophagy will take place as a protective mechanism by degrading the cellular components to prevent spreading of damage in the mitochondrial network.Citation62 The formation of autophagosomes is a consequence of this process.Citation18 Mitophagy plays a potentially protective role in removal of mitochondria damaged by cigarette smoke.Citation63 A cytosolic E3 ubiquitin ligase Parkin and the PTEN-induced putative kinase 1 (PINK1) that is an upstream regulator of Parkin are stimulated in this process.Citation62,Citation64 When the mitochondria are depolarized and fail to regain their membrane potential to protect themselves from detrimental effects, the process of mitophagy begins.Citation62 Recent observations have shown that cigarette smoke exposure induces mitophagy, which contributes to overall mitochondrial quality in the epithelial cells and is likely initiated by PINK1 stabilization. The PINK1–PARK2-mediated pathway may account for mitophagy.Citation65 Knockdown of PINK1 and PARK2 results in enhanced mitochondrial ROS production and cellular senescence in human bronchial epithelial cells, with insufficient mitophagy of the cells. These findings suggest that defective mitochondria in a cell undergo mitophagy in a PINK1-dependent manner and that PINK is the initiator of this process.Citation66
In terms of mechanism (), PINK1/Parkin-mediated mitophagy is a complex process involving ubiquitination and autophagy.Citation67 Upon dissipation of the mitochondrial membrane potential, Parkin translocates to depolarized mitochondria through phosphorylation at Ser65 in the ubiquitin-like domain (Ubl) by PINK1.Citation68 Subsequently, numerous outer mitochondrial membrane proteins are ubiquitinated by Parkin, which in turn triggers accumulation of other proteins on the mitochondrial surface.Citation69,Citation70 In a mechanistic study of the damaged mitochondria, two distinct polyubiquitin chains, linked through K27 and K63 of ubiquitin, were reported to be formed at clustered mitochondria in the presence of Parkin.Citation69 Accompanied with the formation of the specific polyubiquitin chains, a molecular weight shift of voltage-dependent anion channel 1 (VDAC1) seemed to be correlated with an increased level of PINK1, indicating that VDAC1 probably participated in PINK1/Parkin-dependent mitophagy. Indeed, it was identified that phosphorylation of VDAC1 is necessary in Parkin-mediated mitophagy, during which the adapter protein p62 exerts a pivotal role in the clearance of damaged mitochondria since Parkin-dependent mitophagy requires p62/SQSTM1/sequestosome-1 recruitment to the clustered mitochondria for final clearance.Citation70 However, the underlying mechanisms of p62-mediated clearance in this pathway have not been fully understood. Previous studies have indicated that the autophagic clearance was regulated by Ser403 phosphorylation of p62, a step promoted by casein kinase 2 or TANK-binding kinase 1.Citation71,Citation72 Ro et al identified Unc-51-like protein kinase 1 as a novel p62 ser403 kinase, while Sertrin2 is a promoter of Ser403 phosphorylation.Citation73 Recently, multiple phosphorylation sites of p62 in ubiquitin association domains have been recognized,Citation74 of which phosphorylation of Ser409 by Unc-51-like protein kinase 1 significantly increases the binding affinity of p62 to ubiquitin under proteotoxic conditions. Thus, the phosphorylated p62 linked with the damaged mitochondria by ubiquitin is recognized by the receptors on an autophagosome.
P62 also has an impact on the elimination of toxic proteins. Cigarette smoking-related COPD is known to induce an imbalance in protein homeostasis in the mitochondria, which initiates aggregation of ubiquitinated proteins,Citation75 which in turn promotes the accumulation of PINK1 and leads to mitophagy. Thus, autophagy is responsible for the elimination of these aggregative proteins. The targeting cargo of mitophagy to autophagosomes is recognized by a body of ubiquitin-binding autophagic adaptors (eg, p62/SQSTM1/sequestosome-1), which connect the ubiquitin system and the autophagic mechanism, promoting the binding of autophagic substrates and the small ubiquitin-like modifiers that belong to the autophagy-related 8/LC3 family located on the membrane of autophagosomes.Citation76 Furthermore, the cargo receptor for autophagic degradation, p62, has also been identified as a target gene for the transcription factor Nrf2 and creates a positive feedback loop for oxidative stress by activation of antioxidant response.Citation77 Another study indicated that Nrf2 knockout in the Staphylococcus aureus sepsis model induced dysregulated autophagy of the alveolar cells, which led to accumulation of p62 and increased colocalization of LC3 to mitochondria.Citation78 Thus, Nrf2-dependent mitophagy or p62-targeted phosphorylation may provide a potential novel drug target for oxidative stress-related lung disease. Additionally, recent studies have indicated that mitophagic clearance also aggravates the pathogenesis of COPD. Significant accumulation of polyubiquitinated proteins and p62 was detected in the lungs when exposed to cigarette smoke extract, which accelerated lung aging and COPD-emphysema exacerbations.Citation79 P62 as an impaired autophagy marker showed an increased level in aggresome bodies in the cells exposed to cigarette smoke extract. Recently, PINK-induced mitochondrial instability was found to be correlated with Fbxo15, an ubiquitin E3 ligase component.Citation80 The interaction between PINK1 and Fbxo15 triggers polyubiquitination and proteasomal elimination of cardiolipin synthase, thus impairing the mitochondrial membrane.Citation80 Mitophagy also occurred in emphysema and airway disruption, during which process the Akt ubiquitin E3 ligase mitochondrial E3 ubiquitin protein ligase 1 was identified as a novel mediator associated with cigarette smoking-induced pulmonary endothelial cell death and mitochondrial dysfunction.Citation81 The level of mitochondrial E3 ubiquitin protein ligase 1 was noticeably elevated after exposure to cigarette smoke, potentially contributing to endothelial cell death and dysfunction in the lungs of smokers. Thus, we can presume that the alternative PINK targets or ubiquitin E3 ligase may have a potential application in the therapeutic treatment in COPD, and further investigations are necessary to explore the effects of mitophagy in the pathogenesis of COPD.
Conclusion
Oxidative stress is involved in the pathogenesis and progression of COPD and is also of great importance in the initiation of inflammation. It contributes the most to COPD by disturbing the antioxidant system and the balance of oxidant/antioxidant within the cells, accompanied by an increased risk of comorbid diseases or inflammation. As one of the main producers of ROS, mitochondria undergo a series of changes in morphology. These alterations, such as mitochondrial fission/fusion or mitophagy, are stress reactions in response to excessive oxidation, which provides a train of thought of alleviating partial injuries of cells by targeting certain molecules. Therapies targeting enhancement of antioxidants’ ability seem to be able to reduce COPD exacerbations by modulating the oxidative stress. However, untargeted antioxidants such as vitamin E and NAC lack beneficial effects sometimes or may even worsen the disease. Mitochondria-targeted antioxidants such as Mito-TEMPO have been validated for their effective role in restoring impaired mitophagy and in delaying cellular senescence in Parkin-overexpressing cells. Thus, further research that focuses on mitochondria should be carried out because it may provide important information on the potential therapeutic strategies for the treatment of COPD patients.
Acknowledgments
The authors gratefully acknowledge the financial support for this study provided by the Shanghai Committee of Science and Technology (16401901400), the Health Bureau of Shanghai, China (2011ZJ021) and the State Key Laboratory of Clinical Pharmacology Department of Shanghai General Hospital.
Disclosure
The authors report no conflicts of interest in this work.
References
- KoulPAChronic obstructive pulmonary disease: Indian guidelines and the road aheadLung India201330317517724049249
- TanrikuluACAbakayAEvliyaogluOPalanciYCoenzyme Q10, copper, zinc, and lipid peroxidation levels in serum of patients with chronic obstructive pulmonary diseaseBiol Trace Elem Res2011143265966721080098
- DolinayTChoiAMRyterSWHeme Oxygenase-1/CO as protective mediators in cigarette smoke-induced lung cell injury and chronic obstructive pulmonary diseaseCurr Pharm Biotechnol201213676977622201606
- VestboJHurdSSAgustiAGGlobal strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summaryAm J Respir Crit Care Med2013187434736522878278
- TrupinLEarnestGSan PedroMThe occupational burden of chronic obstructive pulmonary diseaseEur Respir J200322346246914516136
- MathesonMCBenkeGRavenJBiological dust exposure in the workplace is a risk factor for chronic obstructive pulmonary diseaseThorax200560864565116061705
- BernardoIBozinovskiSVlahosRTargeting oxidant-dependent mechanisms for the treatment of COPD and its comorbiditiesPharmacol Ther2015155607926297673
- GhoorahKDe SoyzaAKunadianVIncreased cardiovascular risk in patients with chronic obstructive pulmonary disease and the potential mechanisms linking the two conditions: a reviewCardiol Rev201321419620223095685
- BialasAJSitarekPMilkowska-DymanowskaJPiotrowskiWJGorskiPThe role of mitochondria and oxidative/antioxidative imbalance in pathobiology of chronic obstructive pulmonary diseaseOxid Med Cell Longev20162016780857628105251
- Puente-MaestuLLazaroATejedorAEffects of exercise on mitochondrial DNA content in skeletal muscle of patients with COPDThorax201166212112721097816
- YakesFMVan HoutenBMitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stressProc Natl Acad Sci U S A19979425145199012815
- BernardKLogsdonNJMiguelVNADPH oxidase 4 (Nox4) suppresses mitochondrial biogenesis and bioenergetics in lung fibroblasts via a nuclear factor erythroid-derived 2-like 2 (Nrf2)-dependent pathwayJ Biol Chem201729273029303828049732
- KonokhovaYSpendiffSJagoeRTFailed upregulation of TFAM protein and mitochondrial DNA in oxidatively deficient fibers of chronic obstructive pulmonary disease locomotor muscleSkelet Muscle201661026893822
- AravamudanBThompsonMAPabelickCMPrakashYSMitochondria in lung diseasesExpert Rev Respir Med20137663164623978003
- AravamudanBKielAFreemanMCigarette smoke-induced mitochondrial fragmentation and dysfunction in human airway smooth muscleAm J Physiol Lung Cell Mol Physiol20143069L840L85424610934
- HoffmannRFZarrintanSBrandenburgSMProlonged cigarette smoke exposure alters mitochondrial structure and function in airway epithelial cellsRespir Res2013149724088173
- KlaunigJEWangZPuXZhouSOxidative stress and oxidative damage in chemical carcinogenesisToxicol Appl Pharmacol20112542869921296097
- KankiTKlionskyDJThe molecular mechanism of mitochondria autophagy in yeastMol Microbiol201075479580020487284
- KalyanaramanBTeaching the basics of redox biology to medical and graduate students: oxidants, antioxidants and disease mechanismsRedox Biol2013124425724024158
- BarbieriESestiliPReactive oxygen species in skeletal muscle signalingJ Signal Transduct20122012e982794
- BarbieriESestiliPReactive oxygen species in skeletal muscle signalingJ Signal Transduct2012201298279422175016
- GriffithBPendyalaSHeckerLLeePJNatarajanVThannickalVJNOX enzymes and pulmonary diseaseAntioxid Redox Signal200911102505251619331546
- PierrouSBrobergPO’DonnellRAExpression of genes involved in oxidative stress responses in airway epithelial cells of smokers with chronic obstructive pulmonary diseaseAm J Respir Crit Care Med2007175657758617158281
- NagaiKBetsuyakuTSuzukiMDual oxidase 1 and 2 expression in airway epithelium of smokers and patients with mild/moderate chronic obstructive pulmonary diseaseAntioxid Redox Signal200810470571418177232
- SchneiderDGanesanSComstockATIncreased cytokine response of rhinovirus-infected airway epithelial cells in chronic obstructive pulmonary diseaseAm J Respir Crit Care Med2010182333234020395558
- PendyalaSGorshkovaIAUsatyukPVRole of Nox4 and Nox2 in hyperoxia-induced reactive oxygen species generation and migration of human lung endothelial cellsAntioxid Redox Signal200911474776418783311
- ZhangXShanPJiangGCohnLLeePJToll-like receptor 4 deficiency causes pulmonary emphysemaJ Clin Invest2006116113050305917053835
- MartynKDFrederickLMvon LoehneysenKDinauerMCKnausUGFunctional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidasesCell Signal2006181698215927447
- PacheJCCarnesecchiSDeffertCNOX-4 is expressed in thickened pulmonary arteries in idiopathic pulmonary fibrosisNat Med2011171313221217672
- XuSChamseddineAHCarrellSMillerFJJrNox4 NADPH oxidase contributes to smooth muscle cell phenotypes associated with unstable atherosclerotic plaquesRedox Biol2014264265024936437
- MilaraJPeiroTSerranoARoflumilast N-oxide inhibits bronchial epithelial to mesenchymal transition induced by cigarette smoke in smokers with COPDPulm Pharmacol Ther201428213814824525294
- HollinsFSutcliffeAGomezEAirway smooth muscle NOX4 is upregulated and modulates ROS generation in COPDRespir Res20161718427435477
- LiuXHaoBMaAHeJLiuXChenJThe expression of NOX4 in smooth muscles of small airway correlates with the disease severity of COPDBiomed Res Int20162016289181027656649
- JastrochMDivakaruniASMookerjeeSTrebergJRBrandMDMitochondrial proton and electron leaksEssays Biochem201047536720533900
- AgrawalAMabalirajanURejuvenating cellular respiration for optimizing respiratory function: targeting mitochondriaAm J Physiol Lung Cell Mol Physiol20163102L103L11326566906
- MinutoliLPuzzoloDRinaldiMROS-Mediated NLRP3 Inflammasome activation in brain, heart, kidney, and testis ischemia/reperfusion injuryOxid Med Cell Longev20162016218302627127546
- ParkJChoiHMinJSMitochondrial dynamics modulate the expression of pro-inflammatory mediators in microglial cellsJ Neurochem2013127222123223815397
- JiangXLZhongPHuangCLHeFFanXMChenXRThe relationship between nutritional status and oxidative stress markers, pulmonary function in patients with stable chronic obstructive pulmonary diseaseZhonghua Jie He He Hu Xi Za Zhi20174014045 Chinese28100361
- van der ToornMRezayatDKauffmanHFLipid-soluble components in cigarette smoke induce mitochondrial production of reactive oxygen species in lung epithelial cellsAm J Physiol Lung Cell Mol Physiol20092971L109L11419411310
- FolzRJGuanJSeldinMFOuryTDEnghildJJCrapoJDMouse extracellular superoxide dismutase: primary structure, tissue-specific gene expression, chromosomal localization, and lung in situ hybridizationAm J Respir Cell Mol Biol19971743934039376114
- GoldkornTFilostoSLung injury and cancer: mechanistic insights into ceramide and EGFR signaling under cigarette smokeAm J Respir Cell Mol Biol201043325926820525802
- ShapiroSDGoldsteinNMHoughtonAMKobayashiDKKelleyDBelaaouajANeutrophil elastase contributes to cigarette smoke-induced emphysema in miceAm J Pathol200316362329233514633606
- MorettoNBertoliniSIadiciccoCCigarette smoke and its component acrolein augment IL-8/CXCL8 mRNA stability via p38 MAPK/MK2 signaling in human pulmonary cellsAm J Physiol Lung Cell Mol Physiol201230310L929L93822983351
- ShapiroSDIngenitoEPThe pathogenesis of chronic obstructive pulmonary disease: advances in the past 100 yearsAm J Respir Cell Mol Biol200532536737215837726
- SchambergerACStaab-WeijnitzCAMise-RacekNEickelbergOCigarette smoke alters primary human bronchial epithelial cell differentiation at the air-liquid interfaceSci Rep20155816325641363
- SongYSKimMSLeeDHOhDKYoonDY15-Hydroxyeicosatetraenoic acid inhibits phorbol-12-myristate-13-acetate-induced MUC5AC expression in NCI-H292 respiratory epithelial cellsJ Microbiol Biotechnol201525558959725649981
- BuluaACSimonAMaddipatiRMitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS)J Exp Med2011208351953321282379
- BallwegKMutzeKKonigshoffMEickelbergOMeinersSCigarette smoke extract affects mitochondrial function in alveolar epithelial cellsAm J Physiol Lung Cell Mol Physiol201430711L895L90725326581
- OteraHIshiharaNMiharaKNew insights into the function and regulation of mitochondrial fissionBiochim Biophys Acta2013183351256126823434681
- GomesLCDi BenedettoGScorranoLDuring autophagy mitochondria elongate, are spared from degradation and sustain cell viabilityNat Cell Biol201113558959821478857
- EuraYIshiharaNOkaTMiharaKIdentification of a novel protein that regulates mitochondrial fusion by modulating mitofusin (Mfn) protein functionJ Cell Sci2006119Pt 234913492517105775
- ZhangJLiuWLiuJG-protein beta2 subunit interacts with mitofusin 1 to regulate mitochondrial fusionNat Commun2010110120981029
- ZorzanoALiesaMPalacinMMitochondrial dynamics as a bridge between mitochondrial dysfunction and insulin resistanceArch Physiol Biochem2009115111219267277
- OngSBKalkhoranSBCabrera-FuentesHAHausenloyDJMitochondrial fusion and fission proteins as novel therapeutic targets for treating cardiovascular diseaseEur J Pharmacol2015763Pt A10411425987420
- ChenHChomynAChanDCDisruption of fusion results in mitochondrial heterogeneity and dysfunctionJ Biol Chem200528028261852619215899901
- ChenHDetmerSAEwaldAJGriffinEEFraserSEChanDCMitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic developmentJ Cell Biol2003160218920012527753
- SongZChenHFiketMAlexanderCChanDCOPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1LJ Cell Biol2007178574975517709429
- AhmadTSundarIKLernerCAImpaired mitophagy leads to cigarette smoke stress-induced cellular senescence: implications for chronic obstructive pulmonary diseaseFASEB J20152972912292925792665
- KimKYPerkinsGAShimMSDRP1 inhibition rescues retinal ganglion cells and their axons by preserving mitochondrial integrity in a mouse model of glaucomaCell Death Dis20156e183926247724
- WiegmanCHMichaeloudesCHajiGOxidative stress-induced mitochondrial dysfunction drives inflammation and airway smooth muscle remodeling in patients with chronic obstructive pulmonary diseaseJ Allergy Clin Immunol2015136376978025828268
- WangKYanRCooperKFStrichRCyclin C mediates stress-induced mitochondrial fission and apoptosisMol Biol Cell20152661030104325609094
- TwigGShirihaiOSThe interplay between mitochondrial dynamics and mitophagyAntioxid Redox Signal201114101939195121128700
- MizumuraKCloonanSMNakahiraKMitophagy-dependent necroptosis contributes to the pathogenesis of COPDJ Clin Invest201412493987400325083992
- JinSMYouleRJPINK1- and Parkin-mediated mitophagy at a glanceJ Cell Sci2012125Pt 479579922448035
- ItoSArayaJKuritaYPARK2-mediated mitophagy is involved in regulation of HBEC senescence in COPD pathogenesisAutophagy201511354755925714760
- ChenZHKimHPSciurbaFCEgr-1 regulates autophagy in cigarette smoke-induced chronic obstructive pulmonary diseasePLoS One2008310e331618830406
- KageyamaSSouYSUemuraTProteasome dysfunction activates autophagy and the Keap1-Nrf2 pathwayJ Biol Chem201428936249442495525049227
- Shiba-FukushimaKImaiYYoshidaSPINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagySci Rep20122100223256036
- GeislerSHolmstromKMSkujatDPINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1Nat Cell Biol201012211913120098416
- GeggMECooperJMChauKYRojoMSchapiraAHTaanmanJWMitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagyHum Mol Genet201019244861487020871098
- MatsumotoGWadaKOkunoMKurosawaMNukinaNSerine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteinsMol Cell201144227928922017874
- PilliMArko-MensahJPonpuakMTBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturationImmunity201237222323422921120
- RoSHSempleIAParkHSestrin2 promotes Unc-51-like kinase 1 mediated phosphorylation of p62/sequestosome-1FEBS J2014281173816382725040165
- LimJLachenmayerMLWuSProteotoxic stress induces phosphorylation of p62/SQSTM1 by ULK1 to regulate selective autophagic clearance of protein aggregatesPLoS Genet2015112e100498725723488
- TranIJiCNiIMinTTangDVijNRole of cigarette smoke-induced aggresome formation in chronic obstructive pulmonary disease-emphysema pathogenesisAm J Respir Cell Mol Biol201553215917325490051
- LippaiMLowPThe role of the selective adaptor p62 and ubiquitin-like proteins in autophagyBiomed Res Int2014201483270425013806
- JainALamarkTSjottemEp62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcriptionJ Biol Chem201028529225762259120452972
- ChangALUlrichASulimanHBPiantadosiCARedox regulation of mitophagy in the lung during murine Staphylococcus aureus sepsisFree Radic Biol Med20157817918925450328
- VijNChandramaniPWestphalCVHoleRBodasMCigarette smoke induced autophagy-impairment accelerates lung aging, COPD-emphysema exacerbations and pathogenesisAm J Physiol Cell Physiol Epub2016713
- ChenBBCoonTAGlasserJRE3 ligase subunit Fbxo15 and PINK1 kinase regulate cardiolipin synthase 1 stability and mitochondrial function in pneumoniaCell Rep20147247648724703837
- KimSYKimHJParkMKMitochondrial E3 ubiquitin protein ligase 1 mediates cigarette smoke-induced endothelial cell death and dysfunctionAm J Respir Cell Mol Biol201654228429626203915