
Abstract
Introduction
COPD is an inflammatory airway pathology associated with recurrent infection by nontypeable Haemophilus influenzae (NTHi) that is not effectively managed by macrolide antibiotic therapy. We hypothesised that NTHi is able to reside intracellularly within COPD-derived airway epithelial cells (AEC), and that the factors contained in cigarette smoke when coupled with exposure to erythromycin or azithromycin arrest autophagy, the principle mechanism responsible for clearing intracellular bacteria (called “xenophagy”).
Methods
Cultures of bronchial airway epithelial cells derived from control and COPD participants were differentiated at an air–liquid interface and exposed to macrolide antibiotics, 10% cigarette smoke-extract (CSE) and NTHi. Markers of autophagic flux and intracellular NTHi were assessed using Western blot analysis and transmission electron microscopy.
Results
AEC treated with macrolide antibiotics or 10% CSE exhibited a block in autophagic flux as evidenced by a concomitant increase in LC3-II and Sequestosome abundance (vs control; both P < 0.01). While control AEC showed no clear evidence of intracellular NTHi, COPD-derived cultures exhibited abundant NTHi within the cytoplasm. Further, intracellular NTHi that were encapsulated within vesicles propagated from the apical epithelial layer to the basal cell layer.
Discussion
Taken together, our findings indicate that COPD, cigarette smoke and macrolide antibiotics potentiate the susceptibility to persistent intracellular NTHi. A major mechanism for this is arresting normal autophagic flux in airway epithelial cells. Hence, structural modifications that mitigate this off-target effect of macrolides have significant potential to clear intracellular NTHi and thereby reduce the influence of this pathogen in the airways afflicted by COPD.
Introduction
COPD is the third leading cause of death worldwide.Citation1 Alongside consumption of cigarettes, infection with NTHi is a primary factor driving declined lung function and mortality, with approximately 50% of COPD exacerbations attributed to NTHi, which is now a WHO classified priority pathogenic bacterium.Citation1,Citation2 Paradoxically, NTHi is also a clinically benign commensal in the nasopharynx of most healthy individuals.Citation2 Hence, disease-related modifications during COPD produce conditions that strongly favour NTHi infection. Growing evidence for an intracellular aspect to NTHi’s pathogenicity,Citation3 aligns with the pathobiology of COPD, whereby protracted activation and injury of airway epithelial cells (AEC) allows the accumulation of intrinsic defects that promote bacterial invasion and an opportunity to exploit the intracellular environment.
Autophagy is a fundamental intracellular process whereby the autophagosome encapsulates potentially harmful cytoplasmic components and is co-digested with the cargo upon fusion with a lysosome, thus releasing biosynthetic intermediates for metabolic processes (the entire process is termed autophagic flux). This is an essential cellular response to cell stress such as starvation, oxidative stress and inflammation.Citation4,Citation5 Defective autophagic flux has been reported in several chronic lung diseases,Citation6,Citation7 although its role in COPD has not been well defined. However, important for COPD, autophagy is the primary mechanism used to clear invading bacteria, a process called xenophagy.
Xenophagy is a selective form of autophagy that rapidly digests intracellular microbes. Autophagy and xenophagy share molecular mechanisms including mTOR (primary inhibitor of initiation), AuTophaGy (ATG) related complexes that are essential for initiation of auto/xenophagic flux, ATG5, −8 and −16L1 for membrane assembly and maturation, the cargo chaperones Sequestosome, Optineurin, and NDP52, and Light Chain 3-II (LC3-II) which is essential throughout auto/xenophagic flux, LAMP2, RAB7, RAB9, and PtdIns3KC3 (many functions including lysosome formation) and IFN-γ, TLR-3/4, ICAM1, and NOD2 (microbe-host cell interactions).Citation8–Citation10 However, clinically important pathogens have evolved mechanisms allowing them to evade, block and usurp the xenophagic apparatus as an essential aspect of their virulence.Citation11 Some bacteria that infect the airways evade or usurp the xenophagic machinery by secreting inhibitory factors (eg Listeria),Citation1 preventing autophagosome-lysosome fusion (eg Legionella), or upregulating and recruiting LC3 to facilitate their survival and replication (eg S. aureus).Citation10,Citation12 One study found NTHi can exist within late endosomes,Citation13 and more recently NTHi was found to promote the generation of de novo autophagic complexes in Hep-2 cells.Citation14
The prevailing consensus is that autophagic flux is blocked during COPD, which is evidenced by an accumulation of autophagosomes and the inhibition of autophagic survival processes that closely corresponds with COPD-related deficits at the epithelium, including nutrient depletion, fragility, and unscheduled senescence.Citation1,Citation15 Further, exogenous exposures associated with COPD, including cigarette smoke and corticosteroids, are known to block autophagic flux,Citation16 and there is a well-described defect in mitophagy (autophagic clearance of defective mitochondria) that aligns with these observations.Citation17 Hence, in a scenario for COPD in which xenophagic flux is similarly inhibited, an increase in defective xenophagic apparatus could serve to enhance NTHi’s entry into fragile AEC and provide a niche for intracellular persistence. This is consistent with the mechanism employed by S aureus which usurps the xenophagic machinery, where it evades intracellular antimicrobial surveillance and remains metabolically active.Citation11,Citation12
Hence, there is converging evidence that intrinsic defects and exogenous exposures synonymous with COPD potentiate host factors which may restrict xenophagic clearance of NTHi, enabling intracellular residence. Yet, there is a surprising paucity of data describing how the usually innocuous NTHi becomes an influential pathogenic vector in the context of disease, let alone COPD. We report our observations for NTHi within COPD-derived AEC unhindered by autophagic activity, and show AEC exposed to macrolide antibiotics and the factors contained in cigarette smoke exhibit a block in autophagic flux far beyond either treatment in isolation.
Methods
COPD and COPD-Related Exposure Modelling of the Human Airway Epithelium
We employed an air–liquid interface (ALI) model to approximate normal AEC-NTHi interactions and responses to exogenous exposures. This contrasts with prior studies examining NTHi infection, which have primarily utilised undifferentiated and submerged cell line systems, without a disease component or exposure, and which have yielded limited insights. Bronchial brushing for ALI culture samples were approved by the Royal Adelaide Hospital Human Research Ethics (Protocol #R20020811), and all participants provided written informed consent. Cultures were derived from controls (n=5 never smokers, one female, 40 years ±22.3 SD, FEV1/FVC 88.6% ±7) and COPD participants (n=3, one female, 63 years ±4.1 SD, FEV1/FVC 53.1% ±5). Basal progenitor AEC stem cells collected from bronchial brushing samples were propagated at an ALI as previously described.Citation15 Briefly, airway stem cells were expanded in T25 culture flasks, and then transferred to transwells and allowed to propagate to confluence (4–6 days). Thereafter, the apical media was removed and the basal chamber media was replaced with differentiation media containing retinoic acid (24–28 days). Cultures were used as experimental models when the mucociliary phenotype was evident (eg –), via routine light microscopy and when electrical impedance of the epithelial barrier exceeded 500 Ω.cm2. ALI cultures were exposed to a clinical isolate of NTHi (24 h; MOI 50),Citation18 cigarette smoke extract (CSE; as previously described),Citation15 in addition to azithromycin, erythromycin and bafilomycin for 24 h (Merck KGaA Inc, Darmstadt, Germany).
Western Blot Protein Analysis
Protein was isolated from differentiated AEC within the transwells in situ, and Western blot analysis was performed as previously described,Citation19 with the exception of the Rubicon antibody (Cell Signaling Technology Inc., Danvers, MA, USA). Densitometry of histogram analyses was performed using Multi Gauge software (V3.1; Fujifilm, Tokyo, Japan). Density scores were analysed using a random effects gamma (log link) mixed-model regression to allow for correlated treatment responses within each culture. Results were normalised to both β-actin and the biological control and expressed as relative abundance. The statistical analysis was performed using R statistical software (release 3.2.3) for n=4 control cultures.
Electron Microscopy
High-resolution host-microbe and microbe-autophagic apparatus interactions were assessed in situ with previously described methods using transmission electron microscopy (TEM),Citation15 and scanning electron microscopy (SEM).Citation20 For both, the fixative agents were applied directly to cells on the transwell at the end of the exposure period to maintain cell morphology and preserve host–microbe interactions. The transwell membranes were then excised from the transwell frame using a scalpel and embedded/mounted to the TEM mould and SEM peg (respectively), for analysis. TEM and SEM analyses were performed for control (n=3) and COPD (n=3) derived AEC ALI cultures.
Results and Discussion
First, we asked whether COPD-derived AEC exhibit protracted (24 h) intracellular survival of NTHi when compared to cultures derived from control participants. Control AEC cultured with NTHi exhibit strong activation of microbial clearance processes as evidenced by abundant vacuolated structures consistent with lysosomes and autophagosomes (). The relative efficacy of this response meant we were unable to identify intracellular NTHi with certainty, with the exception of irregularly shaped electron dense structures matching the size of NTHi (approximately 500 nm) within double membrane envelopes (), and digestion products within autolysosomes (). However, we cannot rule out that intracellular NTHi was effectively cleared by a related microbial degradative pathway before the 24 h observation interval, and which may be determined by blocking autophagy (eg ATG5 knockout) in control cells using a time course infection protocol. Further, an apparent lack of free NTHi within the cytosol in control AEC may also be explained by effective mucociliary (), barrier and defensin activity, and that stem cells from the relatively older COPD participants (albeit non-significant vs controls), may contribute to intracellular infectivity.
Figure 1 Control-derived airway epithelial cells co-cultured with NTHi exhibit frequent autophagic events, and that may clear invading bacteria before they can be clearly identified within the cytoplasm. Control human air–liquid interface cultures exhibiting mucociliary differentiation and three-dimensional growth characteristics were assessed. (A) An airway epithelial cell (AEC) exhibits active degradative processes after 24 h co-culture with NTHi, and which may represent the final stages of clearing intracellular NTHi. Extracellular NTHi is also observed above the apical aspect of the cell (white arrow). (B) Multiple double membrane structures (white arrows), indicative of autophagosomes containing a circular cargo of similar morphology and size (approx. 500 nm) as NTHi, but which lack the distinct morphology observed for NTHi identified within COPD-derived AEC. Also shown is a double membrane phagophore (double white arrows). (C) Another example of an AEC which exhibits degradative activity. Here autolysosomes are observed at the final stage of a degradative process (white box). (D) The autolysosomes are magnified in D (two are indicated with single white arrows). Further in D, a nearby autophagosome (double white arrow) is also resolved. Data is representative of n=3 control AEC donor cultures.
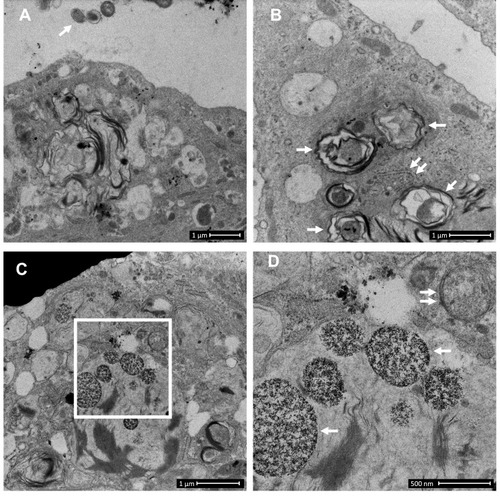
Figure 2 COPD-derived airway epithelial cells and COPD-related exposures exhibit intracellular NTHi and arrested autophagic flux, respectively. Control and COPD human air–liquid interface cultures exhibiting mucociliary differentiation and three-dimensional growth characteristics were assessed. (A–K) Transmission and scanning electron micrographs (TEM/SEM, respectively) of differentiated cultures infected with NTHi. (A and B) Control cells possess abundant cilia (C), villi (V) and apicolateral interaction via tight junctions (TJ). Intracellular NTHi was not observed within AEC after 24 h co-culture (electron dense puncta are cytosolic granules, lysosomes and mitochondria), perhaps due to exclusion from the villi by mucociliary activity/interactions. C and D. COPD-derived cultures associate with NTHi via exposed villi that appear to approximate towards the bacteria (C, dashed box, that is enlarged in H (SEM)), and that are resolved with TEM (D) as membrane projections and pits that interact with NTHi. (E) COPD-derived cultures exhibit NTHi in the cytoplasm (white arrows), that interact with vesicular structures (dashed box). Magnified images for D (I and J) show the NTHi cell wall remains defined while within vesicles. (F) NTHi (white arrows) within proximity to the transwell membrane (TWM) indicates a mode to passage from apical cells to the basal progenitor cell layer. Further, NTHi in vesicles (eg dashed box) which is magnified in (K) remains unaffected by degradative processes in proximity to autophagic activity (solid box). (G) A cluster of NTHi in the basal progenitor cell layer suggests replicative activity, and may indicate an initial stage leading to the formation of an intracellular bacterial community. Discontinuity in the vesicular envelopes for E-F (white chevrons in magnified images (I), (J) and (K) may be caused by NTHi attempting to escape into the cytosol). Data is representative of n=3 COPD and n=3 control AEC donor cultures. (L) Western blot analysis for canonical (LC3-I/II and Sequestosome (Sequest.)) and non-canonical (Rubicon) autophagy factors demonstrate a block in autophagic flux after exposure to 10% cigarette smoke extract (CSE), and the macrolides azithromycin (Az, μg/mL) and erythromycin (Ery, μg/mL). Co-exposure with CSE and either macrolide produces a striking block in flux as evidenced by an increase in LC3-II concomitant with elevation of the autophagy receptor Sequestosome which is normally co-degraded with the cargo during unrestricted autophagy. Exposure to bafilomycin (10 nM) was included as a positive control for the blockade of autophagic flux. Rubicon, an essential component for LC3-Associated Phagocytosis and a negative regulator of canonical autophagy, is moderately upregulated by CSE. (M) Quantitative analysis of protein expression confirms a strong increase in LC3-II and Sequestosome with the combination treatments, that approximates the effect observed for bafilomycin for the Az/CSE exposure. Protein expression is expressed as relative abundance and normalised to the untreated sample and β-actin. Intervals are 95% CI, and results are significant if the confidence intervals do not cross 1, and significant between exposures when CI do not overlap. Exposure period was 24 h for n=4 primary cultures from control individuals.
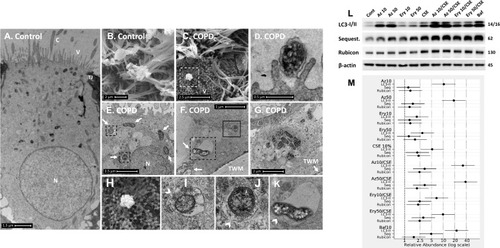
In contrast, COPD-derived AEC were frequently observed interacting with NTHi via villi-like cytoplasmic projections (SEM, , and ), that appear to encapsulate and internalise NTHi (as previously described).Citation21 NTHi were observed in COPD-derived AEC, either free in the cytoplasm or encapsulated within membrane-bound vesicles, possibly endosomes, autophagosome and/or LAPosomes (vesicles of the LC3-Associated Phagocytosis (LAP) non-canonical autophagy pathway,Citation22 –). These observations in COPD-derived AEC support findings for intracellular NTHi identified by Woo et al in the context of otitis media.Citation23 While it is unclear whether an endocytic process is a requisite for NTHi internalisation, we frequently observed an interruption in a NTHi-containing vesicle (NTH-CV), and NTHi in proximity to empty vesicles (––), indicating a potential mechanism of vesicle escape. Indeed, in , NTHi appears to push out from the NTHi-CV, perhaps as part of a replicative process, as evidenced by its elongated, sigmoid, and lobular shape. Clustering depicted in is also suggestive of cell division, and aligns with the observation of an intracellular bacterial community that was recently applied to NTHi in a clinical model of otitis media.Citation24 Interestingly, their close proximity to the transwell membrane () suggests a mode of propagation from the apical to the basal progenitor cell population, which differentiate to generate the three-dimensional epithelial architecture that defines the ALI culture model. Given basal AEC remain viable for an extended period, the ability to traverse the epithelial compartment may partly explain the clinical persistence of NTHi within the COPD airways, whereby exacerbating factors lead to epithelial fragility and senescence that allows a reservoir of NTHi to express virulence factors after a period of host-cell adaption.Citation11 Importantly, we were unable to definitively resolve NTHi within a double membrane bilayer (the diagnostic feature of an autophagosome; eg ), and NTHi-CVs were not observed interacting with lysosomes, even when in proximity to autophagic activity (). As autophagy/xenophagy is usually an efficient process,Citation11 these observations suggest that the inhibition of autophagic flux in the context of COPD may be a primary disease-related phenomena that permits intracellular residence, and resistance to clinical interventions.
The COPD airway is exposed to exogenous factors that modulate autophagy, such as cigarette smoke and corticosteroids,Citation15,Citation16 and which may also reprogram progenitor AEC towards chronic disease.Citation25 Although macrolide therapies are relatively efficacious for the clinical signs of NTHi infection, there is a high rate of reinfection in COPD, and there are macrolide-resistant strains. Further, macrolides are known to modulate autophagy. Rapamycin and bafilomycin have well established pro- and anti-autophagic effects (respectively); however, less is known about the macrolides indicated for respiratory infections. Indeed, of the two studies which examine azithromycin in secondary-cell models of airway disease, one reports an increase in autophagic flux in smooth muscle cells for asthma,Citation26 and the other a decrease in flux and elevated bacterial load in macrophages in the context of cystic fibrosis.Citation27
Hence, we asked whether azithromycin or erythromycin, with or without CSE, can elicit a reduction in autophagy in primary differentiated AEC. We observed a restriction in autophagic flux for either macrolide or CSE-exposed AEC as evidenced by a significant increase in the essential autophagy protein LC3-II (all P<0.05), concomitant with an increase in the autophagy receptor, Sequestosome, which is normally co-degraded with the cargo it chaperones to the autophagosome (–, and see ). While the macrolide-induced elevation in Sequestosome did not reach significance, a reduction of this autophagy receptor would be expected with a significant increase in LC3-II (which should also be degraded during autophagy) if autophagic activity is uninhibited. As expected, CSE elicited a moderate blockade of autophagic flux, with concomitant and significant increases in both LC3-II and Sequestosome. In contrast, the combination of macrolide/CSE resulted in an appreciable block in autophagy, with a significant increase in both LC3-II and Sequestosome for each exposure (consistent with a log-additive effect). The block in autophagy was most pronounced for 50 mg/mL azithromycin/CSE (LC3-II +38.3-fold, CI95% [18.99–77.06], P<0.001; Sequestosome +3.3-fold, CI95% [1.65–6.68], P=0.001), compared to that of bafilomycin (10 nM; LC3-II +22.3-fold, CI95% [11.09–45.00], P<0.001; Sequestosome +4.9-fold, CI95% [2.43–9.86], P<0.001), which is a potent inhibitor of autophagy and a positive control (–). Conversely, while Rubicon was also significantly increased following CSE exposure (P<0.05), its abundance was not substantively influenced by either macrolide, suggesting distinct regulation of LAP vs canonical autophagy in the context of these exposures. LAP is known to suppress canonical autophagy and favour internalisation and infectivity of a number of respiratory pathogens,Citation22 which is consistent with our observations.
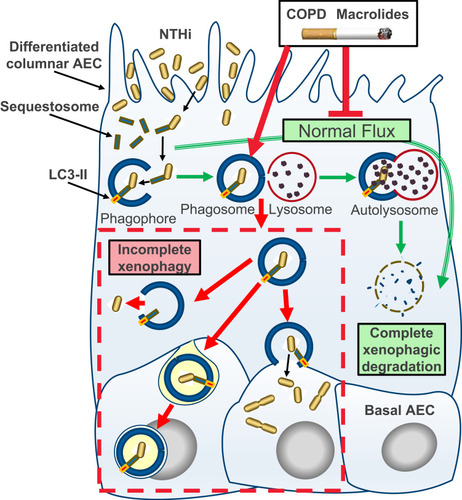
Figure 3 Schematic model of incomplete xenophagy of NTHi in airway epithelial cells (AEC) in the context of COPD and COPD-related exposures. Normally, xenophagy sequesters intracellular bacteria by shuttling invading pathogens using a chaperone protein (eg Sequestosome) to the phagophore, which circularises (phagophore) and docks with a lysosome to form the autolysosome. The autolysosome and cargo are then rapidly co-degraded by low pH and enzymatic activity (“autophagic flux”; green arrows). Intrinsic defects in AEC and exogenous exposures synonymous with COPD including macrolide antibiotics and the factors contained in cigarette smoke inhibit normal autophagic flux and potentiate incomplete xenophagic degradation of intracellular NTHi (red arrows and dashed box). This scenario may allow NTHi to persist intracellularly where it can be released into the cytosol, propagate to the basal cell layer and exhibit metabolic activity consistent with bacterial replication.
Conclusion
For the most part, current therapeutic options for COPD cannot reverse disease progression after smoking cessation, and it is of concern that some therapeutics may contribute to this phenomenon. Here we show that differentiated cultures of COPD-derive primary AEC permit intracellular residence of NTHi (vs control), and that the macrolide antibiotics erythromycin and azithromycin elicit a potent block in autophagic flux, particularly as a co-exposure with CSE (summarised in ). Taken together, our findings raise questions regarding the use of macrolide antibiotics to manage NTHi in current smokers, particularly as the epithelium in these individuals are already accumulating defects that potentiate autophagic insufficiency. An essential next question is whether there exists a causative link consistent with COPD, and COPD-related exposures that arrest autophagic flux, as primary factors that effect a block in xenophagic clearance of intracellular NTHi. So while modification of the antibiotic properties of macrolides to curtail the emergence of resistant strains is a high priority, the additional therapeutic challenge is to address the chemical properties of macrolides that produce off-target effects that limit autophagy.
Disclosure
Lauren O. Bakaletz is the inventor of IP related to the development of a human vaccine antigen that is licensed to GSK and entitled to royalties. Paul N. Reynolds received travel supports from Boehringer Ingelheim, grants from Royal Adelaide Hospital Research Fund, grants from NHMRC, and salary from South Australia Health, during the conduct of the study; grants from NHMRC, outside the submitted work. The authors report no other conflicts of interest in this work.
Acknowledgments
We are grateful for the expertise and support of the clinical staff of the Royal Adelaide Hospital Thoracic Unit and the participants enrolled into this study who generously provided their valuable time and tissue samples. This study was funded by the Royal Adelaide Research Committee, Royal Adelaide Hospital Research Fund, The Health Services Charitable Gifts Board, and the Rebecca L Cooper Medical Research Foundation.
References
- BarnesPJ, BurneyPG, SilvermanEK, et al. Chronic obstructive pulmonary disease. Nat Rev Dis Primers. 2015;1:15076. doi:10.1038/nrdp.2015.7627189863
- BakaletzLO, NovotnyLA. Nontypeable Haemophilus influenzae (NTHi). Trends Microbiol. 2018;26(8):727–728. doi:10.1016/j.tim.2018.05.00129793827
- KingPT, SharmaR. The lung immune response to nontypeable Haemophilus influenzae (lung immunity to NTHi). J Immunol Res. 2015;2015:706376. doi:10.1155/2015/70637626114124
- MartinetW, AgostinisP, VanhoeckeB, DewaeleM, De MeyeretGRY. Autophagy in disease: a double-edged sword with therapeutic potential. Clin Sci. 2009;116(9):697–712. doi:10.1042/CS2008050819323652
- YangJ, CarraS, ZhuWG, KampingaHH. The regulation of the autophagic network and its implications for human disease. Int J Biol Sci. 2013;9(10):1121–1133. doi:10.7150/ijbs.666624339733
- MizumuraK, CloonanS, ChoiME, et al. Autophagy: friend or foe in lung disease? Ann Am Thorac Soc. 2016;13(Suppl 1):S40–7. doi:10.1513/AnnalsATS.201507-450MG27027951
- NakahiraK, Pabon PorrasMA, ChoiAMK. Autophagy in pulmonary diseases. Am J Respir Crit Care Med. 2016;194(10):1196–1207. doi:10.1164/rccm.201512-2468SO27579514
- StaplesKJ, TaylorS, ThomasS, et al. Relationships between mucosal antibodies, non-typeable Haemophilus influenzae (NTHi) infection and airway inflammation in COPD. PLoS One. 2016;11(11):e0167250. doi:10.1371/journal.pone.016725027898728
- BauckmanKA, Owusu-BoaiteyN, MysorekarIU. Selective autophagy: xenophagy. Methods. 2015;75:120–127. doi:10.1016/j.ymeth.2014.12.00525497060
- HuangJ, BrumellJH. Bacteria–autophagy interplay: a battle for survival. Nat Rev Microbiol. 2014;12(2):101–114. doi:10.1038/nrmicro316024384599
- SuiX, LiangX, ChenL, et al. Bacterial xenophagy and its possible role in cancer: a potential antimicrobial strategy for cancer prevention and treatment. Autophagy. 2017;13(2):237–247. doi:10.1080/15548627.2016.125289027924676
- ChandraP, KumarD. Selective autophagy gets more selective: uncoupling of autophagy flux and xenophagy flux in Mycobacterium tuberculosis-infected macrophages. Autophagy. 2016;12(3):608–609. doi:10.1080/15548627.2016.113926327046255
- MoreyP, CanoV, Marti-LliterasP, et al. Evidence for a non-replicative intracellular stage of nontypable Haemophilus influenzae in epithelial cells. Microbiology. 2011;157(1):234–250. doi:10.1099/mic.0.040451-020929955
- Espinoza-Mellado MdelR, Reyes-PicasoC, Garcés-PérezMS, et al. NTHi triggers autophagy in HEp-2 cells. Arch Microbiol. 2016;198(2):199–204. doi:10.1007/s00203-015-1167-326537814
- RoscioliE, TranHB, JersmannH, et al. The uncoupling of autophagy and zinc homeostasis in airway epithelial cells as a fundamental contributor to COPD. Am J Physiol Lung Cell Mol Physiol. 2017;313(3):L453–L65. doi:10.1152/ajplung.00083.201728596293
- WangJ, WangR, WangH, et al. Glucocorticoids suppress antimicrobial autophagy and nitric oxide production and facilitate mycobacterial survival in macrophages. Sci Rep. 2017;7(1):982. doi:10.1038/s41598-017-01174-928428627
- FujiiS, HaraH, ArayaJ, et al. Insufficient autophagy promotes bronchial epithelial cell senescence in chronic obstructive pulmonary disease. Oncoimmunology. 2012;1(5):630–64118/21. doi:10.4161/onci.20297
- PizzuttoSJ, YerkovichST, UphamJW, HalesBJ, ThomasWR, ChangAB. Children with chronic suppurative lung disease have a reduced capacity to synthesize interferon-gamma in vitro in response to non-typeable Haemophilus influenzae. PLoS One. 2014;9(8):e104236. doi:10.1371/journal.pone.010423625111142
- HodgeG, RoscioliE, JersmannH, et al. Steroid resistance in COPD is associated with impaired molecular chaperone Hsp90 expression by pro-inflammatory lymphocytes. Respir Res. 2016;17:135. doi:10.1186/s12931-016-0450-427769261
- HaKR, PsaltisAJ, TanL, WormaldPJ. A sheep model for the study of biofilms in rhinosinusitis. Am J Rhinol. 2007;21(3):339–345. doi:10.2500/ajr.2007.21.303217621821
- KettererMR, ShaoJQ, HornickDB, BuscherB, BandiVK, ApicellaMA. Infection of primary human bronchial epithelial cells by Haemophilus influenzae: macropinocytosis as a mechanism of airway epithelial cell entry. Infect Immun. 1999;67(8):4161–4170. doi:10.1128/IAI.67.8.4161-4170.199910417188
- MostowyS. Autophagy and bacterial clearance: a not so clear picture. Cell Microbiol. 2013;15(3):395–402. doi:10.1111/cmi.1206323121192
- WooJI, OhS, WebsterP, LeeYJ, LimDJ, MoonSK. NOD2/RICK-dependent β-defensin 2 regulation is protective for nontypeable Haemophilus influenzae-induced middle ear infection. PLoS One. 2014;9(3):e90933. doi:10.1371/journal.pone.009093324625812
- HardisonRL, HarrisonA, WallaceRM, et al. Microevolution in response to transient heme-iron restriction enhances intracellular bacterial community development and persistence. PLoS Pathog. 2018;14(10):e1007355. doi:10.1371/journal.ppat.100735530332468
- RahmanSM, JiX, ZimmermanLJ, et al. The airway epithelium undergoes metabolic reprogramming in individuals at high risk for lung cancer. JCI Insight. 2016;1(19):e88814. doi:10.1172/jci.insight.8881427882349
- StamatiouR, ParaskevaE, BoukasK, GourgoulianisKI, MolyvdasPA, HatziefthimiouAA. Azithromycin has an antiproliferative and autophagic effect on airway smooth muscle cells. Eur Respir J. 2009;34(3):721–730. doi:10.1183/09031936.0008940719386688
- RennaM, SchaffnerC, BrownK, et al. Azithromycin blocks autophagy and may predispose cystic fibrosis patients to mycobacterial infection. J Clin Invest. 2011;121(9):3554–3563. doi:10.1172/JCI4609521804191