Abstract
Rationale
The purpose of this study was to investigate the clinical efficacy and safety of a fixed-dose combination of mometasone furoate/formoterol fumarate (MF/F) administered via a metered-dose inhaler in subjects with moderate-to-very severe chronic obstructive pulmonary disease (COPD).
Methods
This multicenter, double-blind, placebo-controlled trial had a 26-week treatment period and a 26-week safety extension. Subjects (n = 1196), at least 40 years old, were current or ex-smokers randomized to twice-daily inhaled MF/F 400/10 μg, MF/F 200/10 μg, MF 400 μg, F 10 μg, or placebo. The trial’s co-primary endpoints were mean changes from baseline, as area under the curve (AUC), in forced expiratory volume (FEV1) over 0–12 hours (AUC0–12 h FEV1) with MF/F versus MF, and in morning (AM) pre-dose (trough) FEV1 with MF/F versus F after 13 weeks of treatment. Key secondary endpoints were the effects of MF/F on respiratory health status using the Saint George’s Respiratory Questionnaire (SGRQ), symptom-free nights, partly stable COPD at 26 weeks, and time to first COPD exacerbation.
Results
The largest improvements in AUC0–12 h FEV1 were observed with MF/F 400/10 μg and MF/F 200/10 μg. Serial spirometry results demonstrated that bronchodilator effects with MF/F occurred rapidly (within 5 minutes), persisted for 12 hours after dosing, and were sustained over the 26-week treatment period. Similar findings were observed for AM pre-dose FEV1, for which effects were further investigated, excluding subjects whose AM FEV1 data were incorrectly collected after 2 days from the last dose of study treatment. Improvements in SGRQ scores surpassed the minimum clinically important difference of more than four units with both MF/F treatments. At 26 weeks, no notable between-treatment differences in the occurrence and nature of adverse events (AEs) were reported. No unexpected AEs were observed. Overall, 90 subjects reported AEs considered to be treatment-related, the most common of which were lenticular opacities, dysphonia, and oral candidiasis.
Discussion
In conclusion, MF/F treatments improved lung function and respiratory health status, reduced exacerbations, and were well tolerated in subjects with moderate-to-very severe COPD.
Introduction
Chronic obstructive pulmonary disease (COPD) is common in adults, with COPD of at least moderate severity affecting an estimated 10% of the world’s population.Citation1 COPD is the third leading cause of death in the US, and is expected to be the third leading cause of death worldwide by 2020.Citation1,Citation2 The burden of COPD on society is large (US$50 billion estimated direct and indirect costs in the US in 2010) and growing. It also poses a tremendous burden on people afflicted with the disease.
COPD is characterized by a progressive decline in lung function and slowly progressing symptoms,Citation3 as described in COPD guidelines. The Global Initiative for Chronic Obstructive Lung Disease (GOLD)Citation4 and a joint statement of the American Thoracic Society (ATS) and European Respiratory Society (ERS)Citation3 provide guidelines on the diagnosis and management of stable COPD. All COPD grades are associated with a postbronchodilator forced expiratory volume (FEV) to forced vital capacity (FVC) ratio ≤0.70, indicating that airway obstruction is only partially reversible. These guidelines define four grades of COPD that stratify severity based on spirometry measurement of FEV in 1 second (FEV1) as a percentage of that predicted. However, many patients with COPD respond significantly, albeit not fully, in response to a bronchodilator.Citation5,Citation6 Although COPD cannot be cured, it is not an untreatable disease.
The GOLD guidelines recommend scheduled maintenance treatment with long-acting bronchodilators for patients with moderate COPD (FEV1 <80% and ≥50% predicted), and also recommend the addition of an inhaled corticosteroid (ICS) to long-acting bronchodilator therapy for patients with severe COPD (FEV1 <50% predicted) and repeated exacerbations. Citation4 In 2011, an official statement of the American College of Physicians (ACP), American College of Chest Physicians (ACCP), ATS, and ERS raised the FEV1 threshold from <50% to <60% predicted for the recommendation of adding an ICS to the bronchodilator regimen of COPD patients who have frequent exacerbations.Citation7 The ACP/ACCP/ATS/ERS guidelines also state that pharmacologic combinations (eg, long-acting β2-agonists [LABA] and ICS) may be used by symptomatic patients with stable COPD and an FEV1 <60% predicted.Citation7
Measurement of FEV1 is a standard endpoint that has been frequently used in pivotal trials evaluating potential COPD treatments.Citation8–Citation11 Other important measures of therapeutic efficacy in COPD treatment studies are patient-centered outcomes, such as improvement of symptoms, exercise tolerance, and/or quality of life. Furthermore, the ability of a COPD treatment(s) to reduce exacerbations, especially those requiring hospitalization, is another widely used endpoint to measure therapeutic value. It is important to note that spirometric responses to treatment, such as improvement in FEV1, do not always correlate with improvements in symptoms, or vice versa.Citation7 This supports a rationale for evaluating multiple efficacy endpoints in clinical trials of COPD treatments.
Fixed-dose ICS/LABA combination inhalers are now available worldwide. Three ICS/LABA combinations are approved for the treatment of COPD: fluticasone propionate/salmeterol (FPS), budesonide/formoterol (BF), and beclomethasone dipropionate/formoterol. FPSCitation8,Citation9,Citation12,Citation13 and BFCitation11,Citation14 have been shown to improve lung function and health status in patients with COPD. Although the existing ICS/LABA components and fixed-dose combinations (FDC) are similar, they are not identical (eg, differences in bronchodilator onsetCitation15 and ICS bioavailabilityCitation16). Accordingly, a new FDC with a different ICS component (mometasone) may bring different features and possible benefits to the maintenance treatment of patients with COPD. Mometasone furoate (MF) and formoterol (F) have been investigated in multiple pharmacologic and clinical studies. MF is distinguished by its high glucocorticoid receptor affinity and potent antiinflammatory activity, in addition to its low systemic bioavailability. Citation17,Citation18 Additionally, F is distinguished by the rapid onset and sustained duration of its bronchodilator effect.Citation19 MF administered via a dry powder inhaler (DPI) has been investigated in COPD.Citation20 F-DPI has been investigatedCitation21,Citation22 and approved for use in COPD. Combined MF/F has been investigatedCitation23–Citation26 and is approved for treatment of patients with asthma, but has yet to be evaluated for the maintenance treatment of patients with COPD.
The objective of the present study was to assess the clinical efficacy and safety of two doses of MF/F metered dose inhaler (MDI) daily: 400/10 μg BID and 200/10 μg BID versus the individual components or placebo in adult subjects with moderate-to-very severe COPD.
Methods
Patients
Included subjects were males or females ≥40 years old with FEV1/FVC ≤0.70, with a postbronchodilator FEV1 of 25%–60% predicted. Additional inclusion criteria were: symptoms of COPD (eg, chronic cough and sputum production not attributable to another disease) for at least 24 months prior to enrollment; current or ex-smokers with ≥10 pack/year history; no use of parenteral steroids, oral steroids, or antibiotics within 4 weeks prior to screening; and clinically acceptable laboratory tests at screening. Female subjects of childbearing potential were required to use a medically acceptable, adequate form of birth control. Subjects were excluded if they had a current diagnosis of asthma, exhibited marked bronchodilator reversibility (increase in FEV1 ≥400 mL) versus baseline pre-bronchodilator FEV1, had a COPD exacerbation within 4 weeks prior to randomization, or required long-term administration of supplemental oxygen (>15 hours/day). Additional exclusion criteria were a history of: lung cancer; alpha-1-antitrypsin deficiency; previous lung surgery; cataract extractions in both eyes; glaucoma or intraocular pressure ≥22 mmHg in either eye; or the presence of clinically significant medical illness(es) that, in the opinion of the principal investigator, could interfere with the study.
Study design
This was a randomized, placebo-controlled, double-blind, double-dummy, multicenter study (ClinicalTrials.gov identifier: NCT0383721) of MF/F 400/10 μg BID and MF/F 200/10 μg BID compared with MF 400 μg BID and F 10 μg BID in adults with moderate-to-very severe COPD. Total dose was delivered after two inhalations BID of the following actuated doses: MF/F 200/5 μg, MF/F 100/5 μg, MF 200 μg, F 5 μg, or placebo. The study was conducted from 2007 to 2010 at 164 centers in North, Central, and South America, Europe, Africa, and Asia. All centers conformed to good clinical practice and to the study protocol. All centers had the protocol approved by an institutional review board and independent ethics committee. Informed consent was obtained from each subject. All subjects completed a 2-week washout/run-in period, in which previous long-acting COPD treatments (LABA, ICS, LABA/ICS FDC, or long-acting anticholinergic [eg, tiotropium]) were discontinued and substituted with an open-label, short-acting β2-agonist (SABA)/short-acting anticholinergic combination.
At baseline, subjects were randomized in a 1:1:1:1:1 ratio to 26 weeks of double-blind treatment with MF/F 400/10 μg BID, MF/F 200/10 μg BID, MF 400 μg BID, F 10 μg BID, or placebo. Comparisons of MF 400 μg and placebo were included, since the clinical effects of the MF-MDI formulation used in this study have not been evaluated previously. All inhalers were MDIs. The active and placebo MF/F and MF inhalers were identical in appearance, as were the active and placebo F inhalers. Spacers were not used in this study.
Efficacy and safety were evaluated over 6 months in the active treatment and placebo groups. The placebo subjects were discontinued from the trial after 6 months, owing to concerns about placebo treatment for a longer period. Seventy-five percent of subjects in each active treatment group were randomly selected to participate in a 26-week safety extension, which began after the initial 26-week treatment period. During the safety extension, FEV1, peak expiratory flow (PEF), and adverse event data, as well as additional data (eg, patient diary, electrocardiogram, COPD stability score, exacerbation evaluation, treatment adherence, and eye examination) were assessed to monitor efficacy and to assure subject safety. In addition, hypothalamic-pituitary-adrenal (HPA) axis and bone mineral density (BMD) data were collected at selected centers at the end of the safety extension (week 52).
Efficacy assessments
This study was designed to evaluate the contribution of each component of a combination inhaler (mometasone plus formoterol formulation) to COPD maintenance treatment. The co-primary endpoints were: 1) MF/F 400/10 μg compared with MF 400 μg for FEV1 area under the curve from 0 to 12 hours post-dose (AUC0–12 h) at the week 13 endpoint (last observation carried forward [LOCF]) to assess the added benefit of F on bronchodilation, and 2) MF/F 400/10 μg and MF/F 200/10 μg compared with F 10 μg for AM predose (trough) FEV1 at the week 13 endpoint to assess the added benefit of MF on trough FEV1. Secondary efficacy endpoints included assessment of changes from baseline in FEV1 AUC0–12 h at day 1, weeks 1, 13, 26, and the 26-week endpoint (LOCF), as well as assessment of changes from baseline in trough FEV1 at each visit and at the 26-week endpoint. Serial spirometry tests were performed at day 1, as well as at weeks 1, 13, and 26, which included measuring the pre-dose FEV1 30 minutes and immediately prior to the AM dose, and then at 5, 15, 30 minutes, and 1, 2, 3, 4, 6, 8, 10, 11, and 12 hours post-dose.
The key secondary efficacy endpoints evaluated for the 26-week treatment period were respiratory health status scores, assessed with the St George’s Respiratory Questionnaire (SGRQ);Citation27 COPD symptom-free nights (combined score of 0 upon awakening for wheezing, cough, and difficulty breathing); and, as defined below, partly stable COPD, and time to first mild, moderate, or severe COPD exacerbation. The SGRQ consists of three component scores (symptoms, activity, impact) and a total score, each of which ranges from 0–100. The better SGRQ scores have a lower numeric value. A four-point difference from baseline or placebo is considered the minimum clinically important difference (MCID).Citation28,Citation29 Partly stable COPD was defined as no use of oral steroid rescue medication; no AM or PM COPD weekly average symptom score >2 during at least 7 of 8 weeks; no moderate or severe COPD exacerbations; no unscheduled visits due to COPD worsenings; and/or no study discontinuation due to treatment failure or treatment-related adverse event (AE).
COPD exacerbations were assessed during the screening and treatment periods, as well as during the 26-week safety extension, and were categorized as mild, moderate, or severe. A mild exacerbation was defined as a clinically judged deterioration of COPD symptoms (managed with increased short-acting bronchodilator use: ≥12 inhalations/day of SABA/short-acting anticholinergic, or ≥2 nebulized treatments/day of 2.5 mg SABA/short-acting anticholinergic) on any two consecutive days. A moderate exacerbation was defined as a clinically judged deterioration of COPD with an acute change in symptoms that required antibiotic and/or oral steroid treatment for lower airway disease. A severe exacerbation was defined as a deterioration of COPD that resulted in emergency treatment or hospitalization due to COPD. Data were analyzed for the time to first mild, moderate, or severe COPD exacerbation and for the time to first moderate or severe COPD exacerbation, excluding mild events.
Safety and e-diary assessments
Safety assessments included monitoring of treatment-emergent AEs (ie, those that occurred during randomized treatment), vital signs, oropharyngeal changes, and forearm bruising. AEs were monitored by investigators and may have included the onset of new illness and the exacerbation of pre-existing conditions (eg, COPD). Laboratory assessments, electrocardiography, and ophthalmologic examinations were conducted at screening and at final visit. Chylack Incorporated (Duxbury, MA) provided guidance for ocular examinations and online training to ophthalmologists for Lens Opacities Classification System III (LOCS III) certification. Measurements of BMD and 24-hour plasma cortisol were conducted at selected centers. Dual-energy X-ray absorptiometry (DXA) scans of the lumbar spine, left total femur, and femoral neck were obtained from a subgroup of subjects at selected centers. CCBR-Synarc (Portland, OR) provided centralized analysis of the DXA scans, project management related to DXA, and instrument quality control.
Each patient was given an e-diary (CareFusion Germany 234 GmbH, Höchberg, Germany) with a built-in spirometer to capture PEF, and a self-contained device to record information about medication use, nocturnal awakenings, COPD symptoms, and stability. COPD stability was evaluated with 5- or 6-point scale (0 = best, 4 to 5 = worst), measuring breathlessness, mucus production, chest tightness, cough, interference with personal care, and interference with outdoor activities. All of the scores, except coughing, were based on 5-point scales from 0 to 4. The coughing score was based on a 6-point scale from 0 to 5. Investigators or designated personnel at each site reviewed and downloaded subjects’ e-diary entries.
Statistical analyses
Efficacy analyses and safety summaries were based on the intent-to-treat principle for all randomized subjects, with at least some follow-up information provided. Subjects who discontinued early were not replaced, and AEs occurring up to 30 days after study completion or discontinuation were reported. The target sample size was 1000 subjects (200 subjects per treatment group). It was predetermined that this sample size would be sufficient to detect a difference of 1.2 L/hour between MF/F 400/10 μg BID and MF 400 μg BID (in change from baseline FEV1 AUC0–12 h) with 91% power and a two-sided alpha level of 5% significance, assuming a pooled standard deviation of 3.6 L/hour. A 1.2 L/hour AUC0–12 h converts to an average difference of 100 mL in FEV1 across 12 hours. A difference of this magnitude is considered clinically meaningful in subjects with this severity of COPD. For AM pre-dose FEV1 at the week 13 endpoint, the contribution of the MF 400 μg BID component was expected to be 80 mL for a target treatment difference of 160 mL between MF/F 400/10 μg BID and placebo. This treatment difference could be detected at 93% power with a two-sided alpha level of 4.9%, assuming a pooled standard deviation of 230 mL. The alpha level was adjusted to allow for a nominal penalty of 0.1%.
The first co-primary efficacy endpoint was the mean AUC0–12 h of the change in FEV1 from baseline to the week 13 endpoint, measuring the contribution of F 10 μg BID to the combination. This analysis compared MF/F 400/10 μg BID versus MF 400 μg BID, MF/F 400/10 μg BID versus placebo, and F 10 μg BID versus placebo. All of these comparisons had to be statistically significant at this dose level of MF/F to assess successfully the F contribution at the overall alpha level of 5%. The second co-primary efficacy endpoint for the study was AM pre-dose FEV1 at the week 13 endpoint, measuring the contribution of MF to the combination. This analysis compared MF/F 400/10 μg BID versus F 10 μg BID, MF/F 400/10 μg BID versus placebo, and MF 400 μg BID versus placebo. All of these comparisons had to be statistically significant at this dose level of MF/F for the study to be successful at an adjusted alpha level of 4.9%. The alpha level was adjusted for an interim analysis to allow for a penalty of 0.1%, preserving the overall alpha level of 5% for the evaluation of the MF contribution. The contribution of MF to the MF/F combination was evaluated by analyzing results in subjects whose AM pre-dose FEV1 measurements were obtained in the protocol-defined time period, using values considered as actual trough FEV1 values. In a second analysis, performed post database lock, FEV1 evaluations for each subject performed ≥2 days after the last dose of treatment were excluded, and the week 13 AM pre-dose FEV1 endpoint was recalculated using the last remaining evaluation, as specified in the study protocol.
Responses for the above co-primary endpoints were analyzed using an analysis of covariance (ANCOVA), extracting sources of variation due to treatment, country, smoking status, and baseline. Pairwise comparisons were based on least squares means from the model. After the significance of co-primary endpoint analyses was confirmed, key secondary endpoints were tested sequentially in order to control the overall alpha level of 5%. If significance was not obtained at any point in this process, then all subsequent comparisons were considered to be descriptive. Changes from baseline to the 26-week endpoint in SGRQ total score and COPD symptom-free nights (AM symptoms) were analyzed using the same ANCOVA as specified for the co-primary efficacy variables. Baseline included AM symptoms over the last week before the first dose. The proportion of subjects with partly stable COPD at endpoint (the last 8 weeks of treatment) was analyzed using the Cochran–Mantel–Haenszel test, controlling for smoking status. The time to first mild, moderate, or severe COPD exacerbation and the time to first moderate or severe COPD exacerbation were analyzed over the 26-week treatment period and the 26-week safety extension using the log-rank test for equality of survival (Kaplan–Meier) curves. The effect of smoking status on the survival curves was examined for the 26-week treatment period. The data were analyzed using SAS® software (v 9.1; SAS Institute, Cary, NC).
Results
Subject disposition
A total of 2936 subjects were screened and 1201 were randomized (). Five subjects were excluded from primary efficacy and safety analyses because they were enrolled at multiple sites. Thus, 1196 subjects were randomized for analysis, meeting the target sample size of 1000 subjects. A total of 956 subjects (80%) completed the 26-week double-blind treatment period, whereas 239 subjects (20%) discontinued from the study early. The two most common reasons for discontinuation were subjects not wanting to continue for reasons unrelated to assigned study treatment (n = 52) and treatment-emergent AEs (n = 50). In the placebo group, 67 subjects (28%) did not complete the 26-week treatment period, including eight treatment failures and four subjects lost to follow-up. In the active treatment groups, 15% to 21% of subjects did not complete the treatment period, with lower numbers of treatment failures and losses to follow-up than in the placebo group ().
Figure 1 Subject disposition.
Abbreviations: AE, adverse event; BID, twice daily; F, formoterol; MF, mometasone furoate; MF/F, mometasone furoate/formoterol fixed-dose combination formulation.
Subject demographics and disease characteristics
Treatment groups were well-balanced regarding baseline demographic characteristics with respect to age, race, and sex (). Overall, 75% of subjects were males, about 70% were white, and the mean subject age was about 60 years. The MF/F 400/10 μg group had a higher proportion of current smokers (56%) than the other treatment groups (range: 50%–53%), and a longer mean smoking history (55 pack-years) than the other treatment groups (range: 40–46 pack-years). Also, FEV1 reversibility at screening tended to be higher in the F 10 μg group (10.37%) than it was in the MF/F 400/10 μg and MF/F 200/10 μg groups (8.69% and 8.47%, respectively). The mean post-bronchodilator FEV1 at screening in all groups was between 38% and 40% predicted.
Table 1 Subject demographics and clinical characteristics
Co-primary efficacy variables
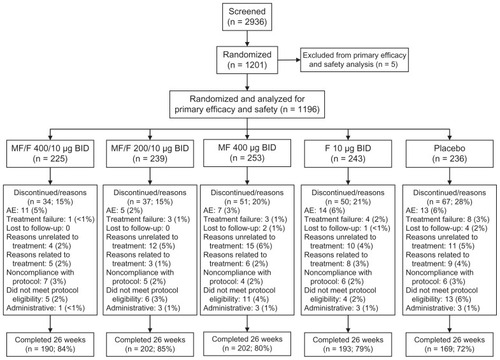
Treatment with MF/F resulted in significant improvements in FEV1, which demonstrated the superiority of the FDC versus the individual components of the combination. At the week 13 endpoint, a significant difference of 126 mL was observed in the mean change from baseline in FEV1 AUC0–12 h between MF/F 400/10 μg and MF 400 μg groups (P < 0.001). MF/F 200/10 μg also significantly improved FEV1 AUC0–12 h versus MF 400 μg (86 mL difference, P < 0.001). A significant improvement of 74 mL was reported for F 10 μg compared with placebo (P = 0.004) (). The effect of MF over 12 hours was evident in the significantly greater improvement with MF 400 μg versus placebo (35 mL, P = 0.038). The significant improvement of FEV1 AUC0–12 h with MF/F 400/10 μg versus F 10 μg (87 mL, P < 0.001) confirms the contribution of MF to the combination.
Figure 2 FEV1 AUC0–12 h at week 13 endpoint (LOCF).
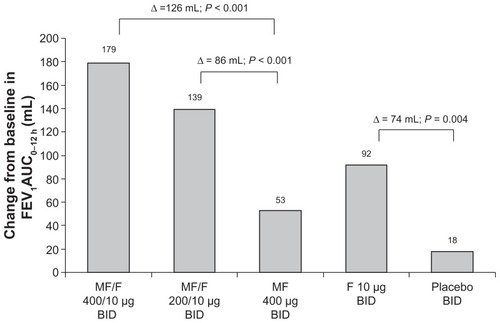
Serial spirometric assessment of FEV1 post-dose at the beginning (day 1) and end (week 26) of treatment identified the rapid onset and sustained duration of bronchodilator effects with MF/F (). Significantly greater increases in FEV1 occurred with both MF/F treatments compared with MF at all time points, including 5 minutes after dosing, both on day 1 (P < 0.001) and at week 26 (P ≤ 0.035). These results show the benefit of F in the combination formulation. The improvements in FEV1 with MF/F at the end of the 26-week treatment period demonstrated the added benefit of the ICS component of the combination. Compared with F 10 μg, MF/F 400/10 μg had significantly greater increases in FEV1 at all time points at week 26 (P ≤ 0.016), whereas MF/F 200/10 μg had significantly greater increases versus F only at the 4- and 8-hour post-dose time points (P ≤ 0.022) Furthermore, as would be expected, both MF/F treatments were superior to placebo (P ≤ 0.019) at all time points during serial spirometry assessments throughout the entire treatment period.
Figure 3 Serial FEV1 post-dose at day 1 (A) and week 26 (B).
Abbreviations: BID, twice daily; FEV1, forced expiratory volume in 1 second; F, formoterol; MF, mometasone furoate; MF/F, mometasone furoate/formoterol fixed-dose combination formulation.
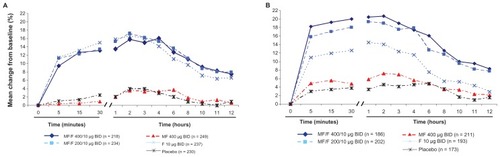
Results with MF/F 400/10 μg and MF/F 200/10 μg versus F 10 μg for the change from baseline in AM pre-dose (trough) FEV1 at the week 13 endpoint indicate a contribution of MF to the combination (). However, the difference between MF/F 400/10 μg and F 10 μg (49 mL) was marginally significant for the pre-specified LOCF analysis (P = 0.062). In analyses for observed cases (analyses based on observations at specified time points, as opposed to observations carried forward for endpoint analyses) at weeks 13 and 26, as well as the week 26 endpoint, statistical significance was achieved, with differences between MF/F 400/10 μg and F 10 μg of 59 mL, 101 mL, and 82 mL, respectively (P ≤ 0.033). A significant mean difference of 101 mL was observed for MF/F 400/10 μg compared with placebo (P < 0.001). Similarly for MF/F 200/10 μg, a significant mean increase of 66 mL was observed compared with placebo (P = 0.013). This supports an overall benefit of both doses of MF/F for AM pre-dose (trough) FEV1, while the nominally increased efficacy of MF/F 400/10 μg over MF/F 200/10 μg provides further evidence of a dose response.
Figure 4 AM pre-dose (trough) FEV1 at week 13 endpoint.
Some subjects had FEV1 measurements long after they had stopped taking the study treatment. In the second analysis of AM pre-dose FEV1, performed after database lock and exclusion of these subjects, statistical significance was achieved for this co-primary endpoint, with a difference of 58 mL between MF/F 400/10 μg and F 10 μg (P = 0.030), and a difference of 105 mL between MF/F 400/10 μg and placebo (P < 0.001).
The co-primary endpoints were analyzed prospectively in all randomized subjects, and post hoc in a subgroup of subjects with baseline FEV1 <50% predicted. The post hoc analyses were performed to assess treatment effects in subjects with severe or very severe COPD. At the week 13 endpoint in this subgroup, significant differences in mean changes from baseline in FEV1 AUC0–12 h occurred, with a difference of 101 mL between MF/F 400/10 μg and MF 400 μg (P < 0.001), 88 mL between MF/F 200/10 μg and MF 400 μg (P < 0.001), and 83 mL between F 10 μg and placebo (P = 0.001). Also at the week 13 end-point, the mean change from baseline in AM pre-dose (trough) FEV1 was 18 mL greater with MF/F 400/10 μg compared with F 10 μg, although the difference was not significant.
Key secondary efficacy variables
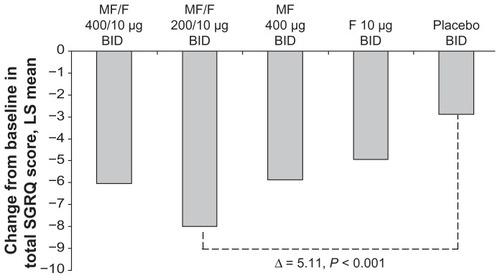
Reported here are changes from baseline in the SGRQ total score. The mean reductions in SGRQ total score at the week 26 endpoint in the MF/F 400/10 μg and placebo groups were 6.04 and 2.88 points, respectively. The difference between these treatments (3.16) was significant (P = 0.020). Also at the week 26 endpoint, the MF/F 200/10 μg group had a mean reduction in SGRQ total score of 7.99 points, with a significant difference (5.11; P < 0.001) from placebo. Both MF/F treatments achieved improvements versus baseline, which surpassed the MCID threshold of greater than a four-unit improvement. The changes from baseline with MF 400 μg (5.87) and F 10 μg (4.93) also surpassed the MCID ().
Figure 5 SGRQ total score change from baseline at week 26 endpoint.
Abbreviations: BID, twice daily; SGRQ, St George’s Respiratory Questionnaire; FEV1, forced expiratory volume in 1 second; F, formoterol; MF, mometasone furoate; MF/F, mometasone furoate/formoterol fixed-dose combination formulation.
The proportion of COPD symptom-free nights over 26 weeks of treatment was highest in the MF/F 200/10 μg group (17%) and lowest in the placebo group (12%). The MF/F 400/10 μg, MF 400 μg, and F 10 μg groups had proportions of 13%, 16%, and 13%, respectively. Comparisons of these treatment differences were not statistically significant. The proportion of subjects with partly stable COPD at week 26 ranged from 37.5% to 43.0% across the treatment groups.
The proportions of subjects who experienced mild, moderate, or severe COPD exacerbations across the 26-week treatment period in the MF/F 400/10 μg, MF/F 200/10 μg, MF 400 μg, F 10 μg, and placebo groups were 37.6%, 32.3%, 33.3%, 40.2%, and 45.7%, respectively. The proportion was significantly lower than placebo for MF/F 400/10 μg (P < 0.027), as well as for MF/F 200/10 μg and MF 400 μg (P < 0.001 and P = 0.003, respectively). In the active treatment groups, the median time to first exacerbation was beyond the 26-week treatment period. Based on the log-rank test for equality of survival curves, MF/F 400/10 μg, MF/F 200/10 μg, and MF 400 μg were superior to placebo (P ≤ 0.027) for all randomized subjects. Similar results were observed for time to first exacerbation in smokers and ex-smokers, for whom log-rank testing of survival curves found MF/F 400/10 μg was superior to placebo (P = 0.027).
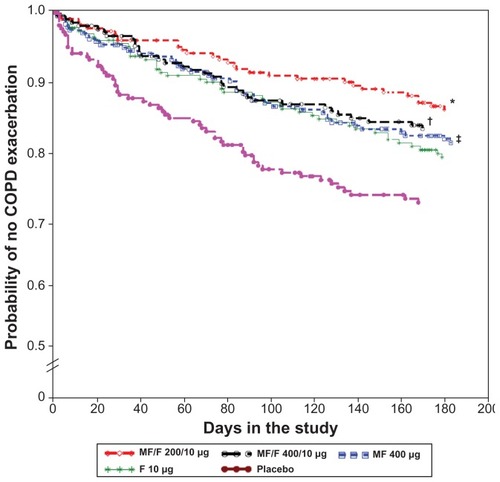
The majority of first COPD exacerbations were mild exacerbations (283/444; 64%). Some subjects on active treatment who experienced a mild exacerbation continued in the study and experienced a moderate or severe exacerbation later on. Therefore, an additional analysis of time to first exacerbation evaluated only subjects whose first event was a moderate or severe exacerbation (). The placebo group had the highest proportion of subjects reporting moderate or severe COPD exacerbations as their first event (24.6%). The proportions of subjects with moderate or severe first exacerbations in the MF/F 400/10 μg, MF/F 200/10 μg, MF 400 μg, and F 10 μg groups were 15.4%, 12.8%, 16.9%, and 18.4%, respectively. Both MF/F groups were superior to placebo (P ≤ 0.006), providing evidence of the effectiveness of both dose levels of MF/F in reducing the incidence of moderate or severe COPD exacerbations.
Figure 6 Time to first moderate or severe exacerbation over the 26-week treatment period.
Abbreviations: F, formoterol; MF, mometasone furoate; MF/F, mometasone furoate/formoterol fixed-dose combination formulation.
Safety
Treatment-emergent adverse events
Both MF/F combination doses were well tolerated during the 26-week treatment period. The overall incidence of treatment-emergent AEs was similar across the treatment groups, ranging from 33.5% for MF/F 200/10 μg to 44.4% for MF/F 400/10 μg (). The percentage of subjects reporting pneumonia (including the AE terms of pneumonia, pneumonia viral, pneumonia aspiration, and lobar pneumonia) during the treatment period was 1.8% overall, and ranged from 0.8% to 3.1% across all treatment groups. If COPD exacerbation met the criteria for a serious AE (eg, was life-threatening, required hospitalization, or prolonged hospitalization), it was recorded as an AE. Serious AEs occurred in 96 subjects (8.0%) during the treatment period, with numbers ranging from 6.7% to 8.9% across groups. A total of 20 subjects (1.7%) had serious AEs considered life-threatening, with numbers similar across all five groups. Overall, 52 subjects (4.3%) discontinued from treatment due to adverse events. A total of 15 (1.3%) subjects died during the treatment period across the five groups; most cases were related to cardiopulmonary events and all were considered unlikely to be related to the study drug. The most commonly reported treatment-emergent AEs during the treatment period were headache (4.4% overall), nasopharyngitis (3.6% overall), upper respiratory tract infection (3.3% overall), COPD (3.1% overall), and hypertension (2.8% overall) (). During the 52-week study period, the incidence of treatment-emergent AEs in the active treatment groups ranged from 44.4% for MF/F 200/10 μg to 51.1% for MF/F 400/10 μg ().
Table 2 Summary of treatment-emergent adverse events
Table 3 Treatment-emergent adverse events in ≥2% of subjects in any treatment group
Treatment-related adverse events
Overall, 90 subjects (7.5%) reported a treatment-related AE, the most frequent of which were lenticular opacities (1 subject MF/F 200/10 μg, 1 subject MF/F 400/10 μg, 2 subjects MF 400 μg, 3 subjects F 10 μg, and 1 subject placebo), dysphonia (2 subjects MF/F 200/10 μg, 1 subject MF/F 400/10 μg, 4 subjects MF 400 μg, and 1 subject placebo), and oral candidiasis, including the AE terms of oral candidiasis, oropharyngeal candidiasis, and oral fungal infection (1 subject MF/F 200/10 μg, 2 subjects MF/F 400/10 μg, 6 subjects MF 400 μg, and 1 subject F 10 μg).
Safety extension
During the 26-week safety extension, the AEs reported by ≥2% of subjects in the active treatment groups were upper respiratory tract infection, nasopharyngitis, headache, COPD, bronchitis, influenza, arthralgia, lenticular opacities, hypertension, and back pain (). During the entire study period (treatment period plus safety extension), 23 subjects (2.4%) reported pneumonia (including the AE terms of pneumonia, pneumonia viral, pneumonia aspiration, and lobar pneumonia) across the four active treatment groups. Thirteen of the 26 events in these 23 subjects were considered to be severe, and all of the events were considered to be unrelated to the study drug.
Table 4 Treatment-emergent adverse events in ≥2% of subjects in any active treatment group over the safety extension
Systemic and ocular effects
No clinically meaningful electrocardiographic changes were observed during the study period, with the exception of three subjects (one in the MF/F 400/10 μg group, one in the MF 400 μg group, and one in the placebo group) who had corrected QT interval increases from baseline. Study treatments had minimal effects on the HPA axis and on BMD, as measured at selected centers over the study period. The treatment groups were well-balanced with regard to baseline 24-hour plasma cortisol (range: 188.9–215.4 μg/dL · hour), and small, insignificant decreases in plasma cortisol were seen across all active treatment groups at weeks 26 and 52. For BMD in the lumbar spine (LS) – the region of greatest interest – decreases in BMD were <2% across all treatment groups at weeks 26 and 52. The greatest loss of LS-BMD was 1.2% in the MF 400 μg group at week 26. The MF/F 200/10 μg group had a slight increase in LS-BMD (0.6%) at week 26, which was significant compared with the MF/F 400/10 μg (−0.9%, P = 0.035), MF 400 μg (−1.2%, P = 0.036), and placebo (−0.007, P = 0.030) groups. Only six subjects had LS-BMD loss >6% during the study period: two subjects each in the MF/F 400/10 μg and MF 400 μg groups, one subject in the MF/F 200/10 μg group, and one subject in the F 10 μg group.
Ophthalmologic examinations found that between 5.0% (MF/F 200/10 μg) and 7.5% (MF 400 μg) of subjects had LOCS III increases of ≥1 unit over the 52-week study period. Four MF/F 200/10 μg subjects and two MF 400 μg subjects reported cataracts and were discontinued from the study, as per protocol. Additionally, intraocular pressure ≥22 mmHg was reported for 14 subjects at week 26, and six subjects at week 52.
Discussion
Treatment for 26 weeks with MF/F 400/10 μg BID and 200/10 μg BID significantly improved lung function and was well tolerated in subjects with moderate-to-very severe COPD. In addition, both MF/F treatments significantly improved respiratory health status as measured by changes from baseline in SGRQ total scores. The magnitude of these changes for both MF/F treatments achieved the threshold for MCID of greater than a four unit improvement from baseline. Also, subjects treated with MF/F experienced significant reductions in the incidence of moderate or severe COPD exacerbations over the 26-week treatment period. The comparatively greater efficacy of MF/F 400/10 μg over MF/F 200/10 μg on lung function suggests a possible dose-response effect of the MF component in the MF/F combination formulation.
Based on 12-hour serial spirometry measurements, MF/F treatment showed a rapid onset of bronchodilation, driven by the F component. The MF/F groups had increases in FEV1 of about 10% at 5 minutes post-dose on day 1, and increases >15% at 5 minutes post-dose at week 26. The latter result shows that the bronchodilator effect of F was maintained over the 26-week treatment period, with no evidence of tachyphylaxis. Furthermore, the 12-hour period for serial spirometry in this study was substantially longer than the 1- or 2-hour serial spirometry assessments in some pivotal trials of other ICS/LABA FDCs,Citation8,Citation9,Citation11 although a recent trial of budesonide/formoterol included serial spirometry data that extended for 12 hours.Citation30
Clinical trials of pharmacotherapy for COPD may fail to show an improvement in SGRQ that meets the four unit MCID for total score.Citation11,Citation14 In the present multicenter trial, both MF/F groups had mean improvements from baseline >4 units, achieving the protocol-defined MCID, while differences from placebo ranged from 3.16 to 5.11 units. The trial demonstrated a clinically relevant improvement in respiratory health status compared with placebo at week 13 and at endpoint.
The probability of a COPD exacerbation was reduced with both MF/F combinations. The MF/F 400/10 μg group had a 17.7% relative risk reduction for mild, moderate, or severe exacerbations, compared with placebo, whereas the MF/F 200/10 μg group had a 29.3% relative risk reduction for these COPD exacerbations. When only the more clinically meaningful moderate or severe COPD exacerbations were analyzed, MF/F 400/10 μg showed a 37.4% relative risk reduction for a moderate or severe exacerbation, whereas MF/F 200/10 μg had a 48.0% relative risk reduction for a moderate or severe exacerbation. MF/F 400/10 μg showed a statistically significant reduction in moderate and severe exacerbations compared not only to placebo but also to MF and F alone. This occurred despite the fact that the treatment period was only 6 months, a time period over which, historically, it has been difficult to show significant effects on exacerbations with pharmacotherapy.
All four active treatments were well tolerated, and there were no notable differences in the occurrence or nature of AEs reported for MF/F 400/10 μg compared with MF 400 μg or F 10 μg alone. The incidence of treatment-emergent pneumonia was low, and no occurrences of pneumonia were considered by investigators to be treatment-related. Regarding systemic safety, effects on HPA axis suppression were quite modest, and no significant demonstrable adverse effects on the cardiovascular system, bone mineral density, lenticular opacities, or intra-ocular pressure occurred in the subpopulation studied. Longer trials would be needed to exclude any long-term effects of MF/F on BMD and ocular safety.
Smoking is the greatest risk factor for COPD, with environmental and occupational exposures contributing as well. Smoking cessation and avoidance of other exposures is an integral part of COPD prevention and treatment. In the present study, the MF/F 400/10 μg group had the greatest proportion of current smokers, as well as a pack-year smoking history that was up to 36% greater than the other treatment groups. The ANCOVA model adjusted for this imbalance by including smoking as a covariate.
The efficacy of an inhaled medication is influenced by factors that include pharmacodynamic properties, particle size, and lung deposition of the drug(s) being administered; the type of inhaler being used (ie, MDI or DPI); as well as the patient’s inhaler technique.Citation31 With HFA-type MDIs, the patient inhales deeply and slowly through the mouth after device actuation to draw medication into the lungs. Proper hand-breath coordination of device actuation and inhalation is necessary to ensure that a sufficient dose of medication is inhaled, and some patients may need to use a spacer or holding chamber to achieve optimal drug delivery. No spacers or holding chambers were utilized in this study. In contrast, drug delivery with a DPI is breath-actuated. The patient places the inhaler in his or her mouth and takes a rapid, deep breath to inhale the medication into the lungs. Deficiencies in a patient’s inhaler technique and treatment adherence can lead to suboptimal outcomes, and thus appropriate device selection and attention to patients’ inhaler acceptance and technique are essential for the successful management of COPD.Citation32,Citation33
Conclusion
Treatment with MF/F was found to be effective for patients with moderate-to-very severe COPD, based on improvement in lung function and health status, as well as reduction in COPD exacerbations. The MF component was shown to contribute to the combination, based on AM pre-dose (trough) FEV1 improvement with MF/F versus F when considering observed cases across all time points. The F component was shown to significantly contribute to the combination, based on FEV1 AUC0–12 h improvement with MF/F versus MF, with improvements in FEV1 that occur rapidly and are sustained over time. The two MF/F combination doses evaluated in this study were well tolerated. Very low rates of pneumonia occurred in all treatment groups.
Principal investigators
Colombia: GA Hincapie Diaz, G Lastra Gonzalez, CE Matiz-Bueno; Costa Rica: A Alvarado Gonzalez, LG Carmona Castro; Ecuador: MI Cherrez Ojeda, JG Tobar Campoverde; Estonia: R Jogi, M Meren, A Press; Guatemala: M Vinicio Flores Belteton, E Contreras Echeverria, JS Guerra Mejia, JM Rodriguez Barillas; Hungary: L Barkai, M Csollak, B Medgyasszay, K Radich, Z Szalai, B Szima, I Vinkler; India: RK Chawla, D Christopher, T Deepak, K Gowrinath, K Jagannath, PG Khatavkar, SK Kochar, S Krishnamurthy, S Kulaprath Rajan, VR Kumar, AK Mani, V Pattabhiraman Ranganathan, KNM Rao, RC Sahoo, MK Thekkinkattil; Mexico: M Acuna, RM Barriga Acevedo, JC Chagoya Bello, DC Hernandez Colin, LA Rendon-Perez, A Dominguez Peregrina, JR Ortiz Peregrina, RQ Zambrano; Netherlands: JPHM Creemers, RALM Stallaert, H Timmer; Peru: A Matsuno Fuchigami, AP Garcia-Calderon, CA Iberico Barrera, JM Rey de Castro Mujica, DJ Salazar Ore, RJ Torres Sales; Philippines: DV Diaz, TS de Guia, C Roa, J Santiaguel; Poland: V Labij, P Kuna, RM Mroz, B Rogala, R Sciborski, E Trebas-Pietras, S Wesolowski, P Wojnowski; Puerto Rico: EA Barranco Santana; Romania: T Mihaescu, D Mocanescu, ID Petrui, C Tanasescu, F Voinea; Russia: J Antonovsky, N Astafieva, B Chernyak, O Krustalev, T Martynenko, V Yakusevich, N Logvinenko, A Zhestkov, O Talibov, A Vizel, N Shaporova, V Trofimov, D Zateyshchikov; Slovakia: G Kosturiakova, H Lescisinova, D Paulovic; South Africa: I Abdullah, FCJ Bester, EJA Biermann, BM Bloy, A Brunning, S Chetty, G Ellis, M Gani, HJ Jansen van Rensburg, G Nieuwoudt, GJ Ras, SJ Schmidt, C Smith, J Theron, AC Wilhase; Sweden: B Lundback; Tunisia: A Chabbou, R Cheikh, A El Kamel, M Jerray, L Megdiche; Ukraine: V Biloglazov, O Dziublyk, Y Feshchenko, I Kaydashev, I Lysenko, Y Mostovyy, S Soldatchenko, V Yef imov, I Lemko, N Shvetz; USA: GH Ayars, M Axler, GW Bensch, SA Brazinsky, EJ Campbell, PM Carter, MA Cromer, EC Cullen, R Fei, RM Gilman, S Garay, G Gwinn, TM Hyers, R Jackson, TD Kaelin, MAR King, T Lee, WM Leeds, G Levinson, EE Lisberg, M Littner, RF Lockey, DG Lorch, TG Moriarty, M Noonan, P Rastogi, A Patel, JL Pearle, F Pozuelo, JD Rossrucker, D Savani, EJ Schenkel, GC Scott, L Sher, TM Siler, S Singh, DM Sinkowitz, CA Smith, R Sockolov, S Spangenthal, JG Sullivan, R Sussman, RN Wolfe; Venezuela: JO Isea Dubuc.
Acknowledgments
Doctors Doherty, Tashkin, and Kerwin were lead investigators on the trial. Doctors Doherty and Tashkin have served as consultants or on advisory boards for Merck & Co, Inc. Doctor Staudinger participated in the study design for the trial. All of the authors reviewed and interpreted the trial data and provided direction and content input for this manuscript. Medical writing and editorial assistance for the preparation of this article was provided by Ken Kauffman, BSc, of AdelphiEden Health Communications, New York, NY. This assistance was funded by Merck Sharp & Dohme Corp, a subsidiary of Merck & Co, Inc, Whitehouse Station, NJ. Editorial assistance was also provided by Jorge Moreno-Cantu, PhD, Global Scientific and Medical Publications, Office of the Chief Medical Officer, Merck Sharp & Dohme Corp. This study was sponsored by Merck Sharp & Dohme Corp.
Disclosures
Dennis E Doherty has served as a consultant for Forest, Ikaria, and Merck; has received research/grant support, via University of Kentucky, from AstraZeneca, Boehringer Ingelheim-Pfizer, Merck, and Novartis; received honoraria from AstraZeneca, Boehringer Ingelheim, Forest, Merck, and Pfizer.
Donald P Tashkin has served as a consultant for AstraZeneca, Boehringer Ingelheim, Dey Laboratories, and Merck; received honoraria from AstraZeneca, Boehringer Ingelheim, and Dey Laboratories; received grants from Almirall, AstraZeneca, Boehringer-Ingelheim, Dey Laboratories, Merck, Novartis, Pfizer, Sepracor, and Forest Laboratories.
Edward Kerwin has received consulting fees from Dey Laboratories, GlaxoSmithKline, MAP Pharma (AstraZeneca), and Sepracor (Sunovion); and received speaking fees from AstraZeneca, GlaxoSmithKline, Merck, and Teva.
Tulin Shekar, Barbara A Knorr, and Heribert Staudinger are employees of Merck Sharp & Dohme Corp. Sibabrata Banerjee is a former employee of Merck Sharp & Dohme Corp.
References
- BuistSCOPD: a common disease that is preventable and treatablePrim Care Respir J2006157916701753
- American College of PhysiciansFour physician organizations issue new clinical recommendations for diagnosing and treating COPD Available at: http://www.acponline.org/pressroom/copd_clinical_recommendations.htmAccessed August 4, 2011
- CelliBRMacNeeWStandards for the diagnosis and treatment of patients with COPD: a summary of the ATS/ERS position paperEur Respir J20042393294615219010
- Global Initiative for Chronic Obstructive Lung Disease (GOLD)Global strategy for the diagnosis, management and prevention of COPD Available at: http://www.goldcopd.org/Accessed July 21, 2011
- DonohueJFTherapeutic responses in asthma and COPD. BronchodilatorsChest2004126125S137S discussion 159S–161S15302773
- TashkinDPCelliBDecramerMBronchodilator responsiveness in patients with COPDEur Respir J20083174275018256071
- QaseemAWiltTJWeinbergerSEDiagnosis and management of stable chronic obstructive pulmonary disease: a clinical practice guideline update from the American College of Physicians, American College of Chest Physicians, American Thoracic Society, and European Respiratory SocietyAnn Intern Med201115517919121810710
- MahlerDAWirePHorstmanDEffectiveness of fluticasone propionate and salmeterol combination delivered via the Diskus device in the treatment of chronic obstructive pulmonary diseaseAm J Respir Crit Care Med20021661084109112379552
- HananiaNADarkenPHorstmanDThe efficacy and safety of fluticasone propionate (250 microg)/salmeterol (50 microg) combined in the Diskus inhaler for the treatment of COPDChest200312483484312970006
- SzafranskiWCukierARamirezAEfficacy and safety of budesonide/formoterol in the management of chronic obstructive pulmonary diseaseEur Respir J200321748112570112
- RennardSITashkinDPMcElhattanJEfficacy and tolerability of budesonide/formoterol in one hydrofluoroalkane pressurized metered-dose inhaler in patients with chronic obstructive pulmonary disease: results from a 1-year randomized controlled clinical trialDrugs20096954956519368417
- VestboJPauwelsRAndersonJAJonesPCalverleyPEarly onset of effect of salmeterol and fluticasone propionate in chronic obstructive pulmonary diseaseThorax20056030130415790985
- BourbeauJChristodoulopoulosPMaltaisFEffect of salmeterol/fluticasone propionate on airway inflammation in COPD: a randomised controlled trialThorax20076293894317557771
- TashkinDPRennardSIMartinPEfficacy and safety of budesonide and formoterol in one pressurized metered-dose inhaler in patients with moderate to very severe chronic obstructive pulmonary disease: results of a 6-month randomized clinical trialDrugs2008681975200018778120
- CoteCPearleJLSharafkhanehASpangenthalSFaster onset of action of formoterol versus salmeterol in patients with chronic obstructive pulmonary disease: a multicenter, randomized studyPulm Pharmacol Ther200922444919071226
- HubnerMHochhausGDerendorfHComparative pharmacology, bioavailability, pharmacokinetics, and pharmacodynamics of inhaled glucocorticosteroidsImmunol Allergy Clin North Am20052546948816054538
- SmithCKreutnerWIn vitro glucocorticoid receptor binding and transcriptional activation by topically active glucocorticoidsArzneim-Forsch1998489569609793625
- AffrimeMBCussFPadhiDBioavailability and metabolism of mometasone furoate following administration by metered-dose and dry-powder inhalers in healthy human volunteersJ Clin Pharmacol2000401227123611075308
- AndersonGPLong acting inhaled beta-adrenoceptor agonists the comparative pharmacology of formoterol and salmeterolAgents Actions Suppl1993432532698103622
- CalverleyPMRennardSNelsonHSOne-year treatment with mometasone furoate in chronic obstructive pulmonary diseaseRespir Res200897319014549
- CazzolaMCentanniSRegordaCOnset of action of single doses of formoterol administered via Turbuhaler in patients with stable COPDPulm Pharmacol Ther200114414511162418
- AalbersRAyresJBackerVFormoterol in patients with chronic obstructive pulmonary disease: a randomized, controlled, 3-month trialEur Respir J20021993694312030736
- MeltzerEOKunaPNolteHNayakASLaforceCon behalf of the P04073 Study InvestigatorsMometasone furoate/formoterol reduces asthma deteriorations and improves lung functionEur Respir J2011 Epub ahead of print at ERJ Express10.1183/09031936.00020310
- NathanRANolteHPearlmanDSTwenty-six-week efficacy and safety study of mometasone furoate/formoterol 200/10 microg combination treatment in patients with persistent asthma previously receiving medium-dose inhaled corticosteroidsAllergy Asthma Proc20103126927920678306
- WeinsteinSFCorrenJMurphyKNolteHWhiteMTwelve-week efficacy and safety study of mometasone furoate/formoterol 200/10 microg and 400/10 microg combination treatments in patients with persistent asthma previously receiving high-dose inhaled corticosteroidsAllergy Asthma Proc20103128028920687982
- MasperoJFNolteHCherrez-OjedaILong-term safety of mometasone furoate/formoterol combination for treatment of patients with persistent asthmaJ Asthma2010471106111520874458
- JonesPWQuirkFHBaveystockCMLittlejohnsPA self-complete measure of health status for chronic airflow limitation. The St George’s Respiratory QuestionnaireAm Rev Respir Dis1992145132113271595997
- JonesPWSt George’s Respiratory Questionnaire: MCIDCOPD20052757917136966
- JonesPWInterpreting thresholds for a clinically significant change in health status in asthma and COPDEur Respir J20021939840411936514
- CelliBRTashkinDPRennardSIMcElhattanJMartinUJBronchodilator responsiveness and onset of effect with budesonide/formoterol pMDI in COPDRespir Med20111051176118821531124
- DolovichMBAhrensRCHessDRDevice selection and outcomes of aerosol therapy: evidence-based guidelines: American College of Chest Physicians/American College of Asthma, Allergy, and ImmunologyChest200512733537115654001
- BarronsRPegramABorriesAInhaler device selection: special considerations in elderly patients with chronic obstructive pulmonary diseaseAm J Health Syst Pharm2011681221123221690428
- MelaniASInhalatory therapy training: a priority challenge for the physicianActa Biomed20077823324518330086