Abstract
Background
The clinical efficacy and safety of a mometasone furoate/formoterol fumarate (MF/F) fixed-dose combination formulation administered via a metered-dose inhaler was investigated in patients with moderate to very severe chronic obstructive pulmonary disease (COPD).
Methods
Two 52-week, multicenter, double-blind, placebo-controlled trials with identical study designs were conducted in current or ex-smokers (aged ≥40 years), and pooled study results are presented herein. Subjects (n = 2251) were randomized to 26 weeks of twice-daily treatment with MF/F 400/10 μg, MF/F 200/10 μg, MF 400 μg, F 10 μg, or placebo. After the 26-week treatment period, placebo subjects completed the trial and 75% of subjects on active treatment entered a 26-week safety extension. Coprimary efficacy variables were mean changes in forced expiratory volume in one second (FEV1), area under the curve from 0 to 12 hours postdose (AUC0–12 h), and morning predose/trough FEV1 from baseline to the week 13 endpoint. Key secondary efficacy variables were St George’s Respiratory Questionnaire scores, symptom-free nights, time-to-first exacerbation, and partly stable COPD at the week 26 endpoint.
Results
In the 26-week treatment period, significantly greater increases in FEV1 AUC0–12 h occurred with MF/F 400/10 versus MF 400 and placebo at the week 13 and week 26 endpoints (P ≤ 0.032). These increases were over three-fold greater with MF/F 400/10 than with MF 400. Also, significantly greater increases in morning predose/trough FEV1 occurred with MF/F 400/10 versus F 10 and placebo at the week 13 endpoint (P < 0.05). The increase was four-fold greater with MF/F 400/10 than with F 10. All active treatment groups achieved minimum clinically important differences from baseline (>4 units) in St George’s Respiratory Questionnaire scores at week 26. Symptom-free nights increased by ≥14% in the MF/F 400/10, MF 400, and F 10 groups (P ≤ 0.033 versus placebo). The incidence of exacerbations was lower in the MF/F groups (≤33.3%) than it was in the MF, formoterol, and placebo groups (≥33.8%) over the 26-week treatment period. The incidence of adverse events was similar in the active-treated and placebo-treated subjects across 26 weeks of treatment. Over the 1-year study period, there were no notable differences in the incidence or types of adverse events between the MF/F 400/10 and MF/F 200/10 groups compared with the MF or formoterol groups. Differences in rates of individual treatment-emergent adverse events were <3% between treatment groups. Rates of pneumonia were low (≤2%) across all treatment groups.
Conclusion
Patients treated with MF/F demonstrated significant improvements in lung function, health status, and exacerbation rates. Although significant improvements were seen with both doses, a trend showing a dose-response effect was observed in the lung function measurements.
Introduction
Chronic obstructive pulmonary disease (COPD) is a chronic and progressive disease that has an enormous impact on patient health and health care resources. In the US, COPD affects more than 5% of the adult population and is the third leading cause of death. Recent estimates of total economic costs in the US are approximately $50 billion, with the direct cost of medical care approaching $30 billion.Citation1 COPD is usually characterized by dyspnea and chronic productive cough,Citation2 airflow obstruction that is not fully reversible, and a heightened inflammatory process in the lungs.Citation2 Recurrent exacerbations, a hallmark of unstable COPD, further impair quality of life, accelerate disease progression,Citation2 and account for more than 70% of the economic burden (direct costs) of COPD on the health care system.Citation2
Recent guidelines published by the Global Initiative for Chronic Obstructive Lung Disease (GOLD) list potential treatment goals, ie, relieving symptoms, preventing progression, improving exercise tolerance and health status, preventing and treating exacerbations and complications, and reducing mortality.Citation2 Bronchodilators (anticholinergics, β2-agonists, and methylxanthines) relax airway smooth muscle and improve lung emptying during tidal breathing at rest and during exertion.Citation3 Long-acting inhaled bronchodilators, consisting of the long-acting anticholinergic, tiotropium, and long-acting β2-agonists (LABAs), including twice-daily salmeterol and formoterol, are recommended as monotherapy for patients with moderate to severe COPD,Citation2 and have been shown to improve lung function, decrease symptoms, and reduce exacerbations.Citation4–Citation8 In clinical trials, formoterol has demonstrated a more rapid onset of bronchodilation compared with salmeterol in patients with COPD.Citation9
For patients with more advanced COPD who continue to have repeated exacerbations despite maximized use of bronchodilators, the addition of an inhaled corticosteroid is recommended.Citation2 Inhaled corticosteroids have been shown to increase the forced expiratory volume in one second (FEV1) when added to the maintenance regimen of severe COPD patientsCitation10,Citation11 and to reduce the severityCitation12 and frequency of exacerbations.Citation11 Mometasone furoate (MF) in a dry-powder inhaler administered at a total daily dose of 800 μg for 12 months demonstrated significant improvements compared with placebo in lung function, time-to-first exacerbation, number of exacerbations, symptoms, and health status.Citation13 However, inhaled corticosteroid monotherapy is not recommended in COPD treatment guidelines, and inhaled corticosteroids are not approved for the treatment of COPD in the US. It is when inhaled corticosteroids are combined with LABAs that their effects become significant.
Concomitant therapy with an inhaled corticosteroid plus LABA is recommended for patients with moderate to severe COPD. Recent guidelines from GOLDCitation2 and the National Institute for Clinical ExcellenceCitation14 recommend inhaled corticosteroid/LABA combination treatment for patients with FEV1 <50% predicted, while a threshold of <60% predicted for use was recommended by a joint position paper of the American Thoracic Society/European Respiratory Society.Citation1 Three inhaled corticosteroid/LABA combination products, ie, fluticasone propionate/salmeterol,Citation15–Citation17 budesonide/formoterol,Citation18,Citation19 and beclomethasone dipropionate/formoterol,Citation20,Citation21 have been shown to improve lung function and health status in patients with COPD and are approved by some regulatory agencies for the maintenance treatment of COPD. Mometasone furoate/formoterol fumarate (MF/F) was recently approved for the treatment of asthma, and two Phase III trials assessing the effect of MF/F in subjects with severe to very severe COPD symptoms were recently completed. We present pooled results from the two Phase III trials of MF/F in subjects with moderate to very severe COPD, which have been published individually, and present results in more detail.
Pooling results from these two studies of identical design improves the precision of the estimated treatment effect for MF/F, and provides a larger safety database for evaluation. Pooling also gives additional information on time-to-first COPD exacerbation, given that exacerbations occur episodically and often require larger study populations and extended follow-up times to discern treatment benefits on exacerbation rates.
Methods
The two studies were of identical design. These randomized, placebo-controlled, double-blind, double-dummy, multicenter Phase III trials evaluated twice-daily treatment with MF/F 400/10, MF/F 200/10, MF 400, F 10, and placebo in adults at least 40 years of age, with moderate to very severe COPD. All treatments were administered via pressurized metered-dose inhaler. Subjects were enrolled at mutually exclusive centers across North, Central, and South America; Europe; Africa; and Asia (131 centers in one trial and 164 centers in the other trial). Both trials were registered on ClinicalTrials.gov (identifier numbers NCT0383435 and NCT0383721). A prospective statistical analysis plan for evaluation of pooled results was completed before unblinding of the two studies.
The studies were conducted in accordance with the principles of Good Clinical Practice and were approved by the appropriate institutional review boards and regulatory agencies. All patients gave written informed consent before enrollment. All subjects were monitored for COPD exacerbations and were provided with a COPD action plan with immediate availability of emergency rescue medication.
Subjects were ex-smokers or current smokers with a smoking history of ≥10 pack-years and had symptoms of COPD for at least 24 months. For inclusion, subjects were required to have a diagnosis of moderate to very severe COPD (based on a prebronchodilator FEV1/forced vital capacity [FVC] ratio of ≤70%), and post-bronchodilator FEV1 between 25% and 60% predicted normal at the screening visit. Subjects who experienced an increase in absolute FEV1 of ≥400 mL at the screening visit or prior to the baseline visit within 30 minutes after administration of four inhalations of albuterol (salbutamol) 360–400 μg total, or nebulized 2.5 mg albuterol (salbutamol) were not enrolled. Specific exclusion criteria were current diagnosis of asthma, oxygen dependence, significant ocular disease (eg, cataracts, glaucoma), abnormal bone density scan, visible evidence of oropharyngeal candidiasis, COPD exacerbation requiring medical intervention within 4 weeks of randomization, oral or parenteral corticosteroid use within 6 weeks of screening, and any clinically significant medical disorder.
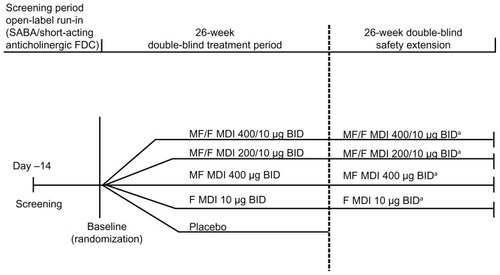
Screening was followed by a 2-week, open-label run-in period, in which short-acting β2-agonist (SABA)/short-acting anticholinergic fixed-dose combination treatment was provided for use as needed. At the baseline visit, subjects who qualified were randomized in a 1:1:1:1:1 ratio to twice-daily treatment with MF/F 400/10 μg, MF/F 200/10 μg, MF 400 μg, F 10 μg, or placebo for 26 weeks (treatment period). Total doses were delivered after two inhalations twice daily of the following actuated doses: MF/F 200/5 μg, MF/F 100/5 μg, MF 200 μg, F 5 μg, or placebo. The number of inhalations and the treatment schedule were identical in each arm, for a total of four inhalations (two inhalations from each of two separate devices) twice daily. At the end of the treatment period, 75% of subjects in each active treatment group were randomly selected to continue their current treatment, in a double-blind fashion, for an additional 26 weeks (safety extension, ).
Figure 1 Study design.
a75% of each group were randomly selected to continue into the safety extension.
Abbreviations: BID, twice daily; F, formoterol; FDC, fixed-dose combination; MDI, metered-dose inhaler; MF, mometasone furoate; MF/F, mometasone furoate/formoterol fumarate fixed-dose combination formulation; SABA, short-acting β2-agonist.
Efficacy assessments
Clinical visits occurred at screening, baseline, day 1, weeks 1, 4, 13, 26, 39, and 52, and/or end of treatment (generally defined as the last week of treatment for each subject). Efficacy was evaluated by pulmonary function tests at all visits, and serial spirometry was performed at baseline, on day 1, and at weeks 1, 13, and 26. Subjects were contacted by telephone the day before each visit and reminded of restricted medication washout times before the visit. Study-provided SABA metered-dose inhalers and nebulizations of 2.5 mg albuterol (salbutamol) were to be withheld for at least 4 hours before visits. Washout times for vaccines (eg, influenza and hepatitis) and immunotherapy were one week and 24 hours, respectively. Investigators attempted to use one spirometer consistently on each subject, and the spirometer was calibrated at each visit. The investigator or qualified designee obtained three FEV1 and three FVC measurements at each visit that met the American Thoracic Society/European Respiratory Society guidelines for test acceptability and reproducibility.Citation22 Study sites used a centralized data system (MasterScope CT) to capture spirometry results at each visit. Spirometry was performed approximately 12 hours after the last dose of study medication.
Patients recorded rescue medication usage (short-acting β2-agonist/short-acting anticholinergic), oral prednisone/prednisolone use, number of nocturnal awakenings requiring rescue medication, peak expiratory flow measurements, and morning and evening COPD symptom scores daily in e-diaries. Pulmonary health status was assessed using the St George’s Respiratory Questionnaire (SGRQ). Deteriorations of COPD symptoms were recorded as exacerbations and classified according to severity, ie, mild (managed with increased short-acting bronchodilator use on any two consecutive days), moderate (required antibiotic and/or oral steroid treatment), or severe (resulted in emergency treatment or hospitalization).
Primary and secondary endpoints were the same in both studies: to assess the contribution of formoterol, the first coprimary endpoint was mean FEV1 from 0 to 12 hours postdose (AUC0–12 h) of the change from baseline to the week 13 endpoint. Comparators were MF/F 400/10 versus MF 400, MF/F 400/10 versus placebo, and F 10 versus placebo. Baseline FEV1 was the average of the two predose/trough FEV1 measurements (30 minutes before dosing and immediately before dosing) at the baseline visit. To assess the contribution of MF, the second coprimary endpoint was the mean change from baseline to the week 13 endpoint in morning predose/trough FEV1. Comparators were MF/F 400/10 versus F 10, MF/F 400/10 versus placebo, and MF 400 versus placebo.
Key secondary efficacy outcomes included change from baseline in SGRQ, time-to-first COPD exacerbation, proportion of COPD symptom-free nights, and partly stable COPD. The SGRQ is a three-component questionnaire that measures symptoms, activity, and social and psychological impacts.Citation23 Total score ranges from 0–100, with a higher score indicating greater disease burden. A difference in total score of 4 points, either from baseline or between treatment groups, has been established as the minimum clinically important difference.Citation24 A symptom-free night was defined as a combined score of 0 upon awakening, prior to the use of study drug or rescue medication, across three domains, ie, wheezing, cough, and difficulty breathing. Exacerbation data were analyzed for the time-to-first mild, moderate, or severe COPD exacerbation and for the time-to-first moderate or severe COPD exacerbation, excluding mild events. The statistical analysis plan prespecified analysis of time-to-first mild, moderate, or severe exacerbation, whereas analysis of time-to-first moderate or severe exacerbation was planned after finalization of the statistical analysis plan but before unblinding of the database. Partly stable COPD was a composite measure of the following outcomes: no use of oral steroid rescue medication; no morning or evening COPD weekly average symptom score greater than 2 during at least 7 of 8 weeks; no moderate or severe COPD exacerbations; no unscheduled visits due to COPD worsening; and no study discontinuation due to treatment failure or treatment-related adverse event. Secondary outcomes were evaluated over the 26-week treatment period.
Safety assessments
Subjects at all centers were monitored at each visit for treatment-emergent adverse events, vital signs, medication use, oropharyngeal changes, and forearm bruising. Ophthalmic examinations (measurement of intraocular pressure and Lens Opacities Classification System, Version III [LOCS III] assessments of cataracts and lens opacification), hypothalamic-pituitary-adrenal axis assessments (24-hour plasma cortisol, at selected centers), and bone mineral density measurements (lumbar spine, left total femur, and femoral neck, at selected centers), were performed at the beginning of the trial and at week 26 and week 52.
Statistical analysis
Statistical analyses were carried out using SAS® software (v 9.1; SAS Institute, Cary, NC). The total target sample size for analysis of pooled results was planned for 2000 patients (400 per treatment group). This sample size allows detection of a difference of 0.8 L × hour between MF/F 400/10 and MF 400 in change from baseline FEV1 AUC0–12 h at the week 13 endpoint, with 90% power and a two-sided alpha level of 5% significance, assuming a pooled standard deviation of 3.6 L × hour. An 0.8 L × hour AUC converts to an average difference of 67 mL in FEV1 across a 12-hour time period. For the morning predose/trough FEV1 at the week 13 endpoint, the contribution of the MF 400 component was expected to be about 53 mL. This treatment difference can be detected at a power of 90% with a two-sided alpha level of 5%, assuming a pooled standard deviation of 230 mL.
Part of the overall effect examination was to compare the MF/F 200/10 and 400/10 μg doses and identify the possible added benefit of the higher dose of MF/F. Pooling the studies provided greater precision to the effect of each dose so such a comparison could be made. Furthermore, pooling the studies doubled the sample size and increased the power to detect treatment differences.
An analysis of covariance, extracting sources of variation due to treatment, country, smoking status, and baseline as covariates, was used to analyze responses for the change from baseline of the FEV1 AUC0–12 h and the morning predose/trough FEV1. Pairwise comparisons were based on least squares means from the model. An analysis of variance, extracting sources of variation due to treatment, country, and smoking status, was performed as a confirmatory analysis for these treatment comparisons. Following testing of the coprimary endpoints at a given dose level of MF/F, tests for the key secondary endpoints at the given dose level were performed sequentially versus placebo. Changes from baseline to the 26-week endpoint (last observation carried forward) in SGRQ total score and the proportion of COPD symptom-free nights, were analyzed using the same analysis of covariance as specified for the lung function coprimary efficacy variables.
For the time-to-first mild, moderate, or severe COPD exacerbation, the log-rank test for equality of survival curves was used with smoking and study as covariates. A GENMOD model was applied to adjust for smoking and study, and assumed a negative binomial distribution of events. Kaplan–Meier curves were used to display these treatment responses. In addition, the effect of smoking status (current versus former) on the survival curves was examined. Assessments were repeated for the lower dose (MF/F 200/10 μg). Hazard ratios were calculated for each active treatment versus placebo on two endpoint evaluations over 26 weeks, ie, time-to-first mild, moderate, or severe exacerbation, and time-to-first moderate or severe exacerbation. However, this required a separate analysis using the Cox proportional hazards method, which used the same covariates (smoking and study) as the log-rank test.
Results
Subject disposition and demographics
Of 5249 subjects screened, 2251 were randomized to treatment. Nine subjects were enrolled at two sites simultaneously and were excluded from analysis. Five subjects were randomized but did not receive study drug. A total of 1796 subjects completed the treatment period. The primary reasons for discontinuation were adverse events (4%) and subjects not wishing to continue for reasons unrelated to the assigned treatment (5%, ).
Table 1 Disposition of patients following randomized treatment assignment: number (%) of patients during the treatment period
Demographic characteristics and baseline lung function are presented in . Overall, 76% of subjects were males, 72% were white, and mean age was approximately 60 years. Demographics and disease characteristics were generally well balanced between treatment arms. However, the MF/F 400/10 group had a higher smoking burden than the other treatment groups (51% current smokers; 47 pack-years mean smoking history).
Table 2 Summary of demographic data and baseline characteristics
Coprimary efficacy outcomes
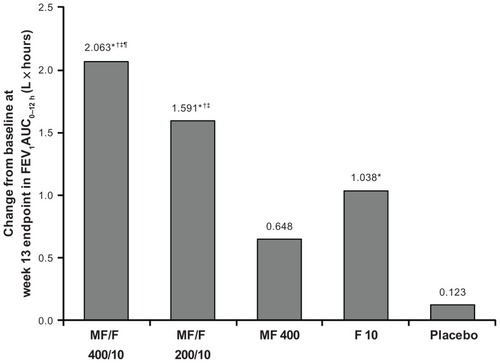
Improvements in FEV1 AUC0–12 h were significantly greater for MF/F 400/10 compared with MF 400 (primary comparison; P < 0.001) and placebo (primary comparison; P < 0.001) at the week 13 primary endpoint () and all other endpoints. F 10 was also significantly superior to placebo (primary comparison) at all endpoints (P ≤ 0.008 for all). These results demonstrate the significant contribution of formoterol. Improvements in FEV1 AUC0–12 h were also significantly greater for MF/F 200/10 compared with MF 400 and placebo at all time points (P < 0.001 for all). Both doses of MF/F were significantly superior to F 10 at almost all endpoints, supporting the contribution of MF to the combination (the difference between MF/F 200/10 and F 10 was not significant at day 1). Improvements in FEV1 AUC0–12 h were significantly greater for 400/10 compared with 200/10 at the week 13 endpoint (P = 0.031) and at 26 weeks (P < 0.05), demonstrating a dose-response relationship. Changes from baseline in FEV1 AUC0–12 h are shown as standardized FEV1 values in liters (FEV1 AUC0–12 h divided by 12) in .
Table 3 Change from baseline in FEV1 AUC0–12 h in all randomized subjects
Figure 2 Change from baseline in FEV1 AUC0–12 h (L × hour) at week 13 (last observation carried forward).
Abbreviations: AUC0–12 h, area under the curve from 0 to 12 h postdose; FEV1, forced expiratory volume in 1 second; F, formoterol; MF, mometasone furoate; MF/F, mometasone furoate/formoterol fumarate fixed-dose combination formulation.
Serial spirometry data
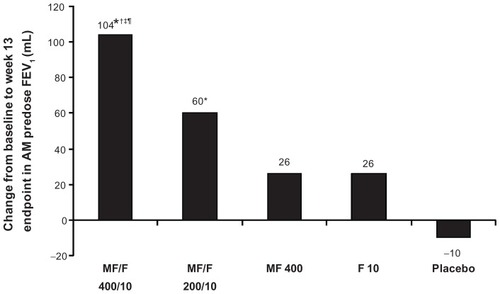
Improvements in AM predose/trough FEV1 were significantly greater for MF/F 400/10 compared with F 10 (primary comparison; P ≤ 0.008) and placebo (primary comparison; P < 0.001) at the week 13 primary endpoint and at all other time points (eg, week 26), supporting the contribution of MF to the combination (). No significant difference was observed between MF 400 and placebo (primary comparison) for change in morning predose/trough FEV1 at 13 weeks ().
Table 4 Change from baseline in morning predose/trough FEV1 in all randomized subjects
Figure 3 Morning predose/trough FEV1 at week 13 endpoint (last observation carried forward).
Abbreviations: FEV1, forced expiratory volume in 1 second; F, formoterol; MF, mometasone furoate; MF/F, mometasone furoate/formoterol fumarate fixed-dose combination formulation.
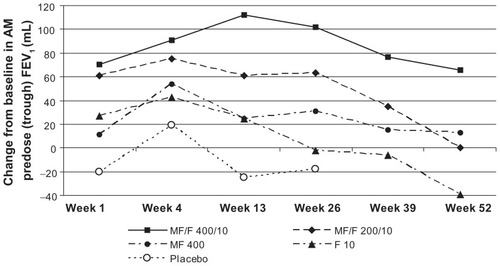
The increase in morning predose/trough FEV1 was not significantly greater with MF/F 200/10 compared with F 10 at the week 13 endpoint. However, MF/F 200/10 was significantly superior to placebo at all endpoints. MF/F 400/10 was significantly superior to MF/F 200/10 at the week 13 endpoint, consistent with the dose-response relationship seen in the analysis of FEV1 AUC0–12 h. MF/F 400/10 was also significantly superior to MF 400 at the week 13 endpoint. No significant improvement was seen when comparing MF/F 200/10 and MF 400 ().
Figure 4 Changes from baseline in morning predose/trough FEV1 over the study period (last observation carried forward).
Abbreviations: F, formoterol; FEV1, forced expiratory volume in 1 second; MF, mometasone furoate; MF/F, mometasone furoate/formoterol fixed-dose combination formulation.
Secondary assessments
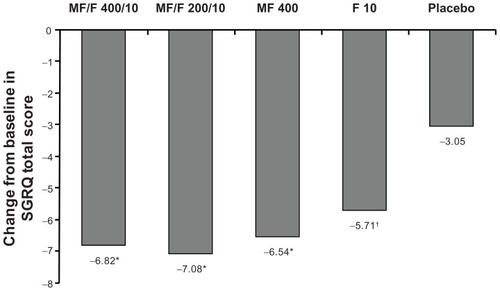
Baseline mean total SGRQ scores were similar between groups (range 45.6–47.6). At the week 26 endpoint, all active treatment arms resulted in a clinically meaningful improvement in mean total SGRQ score of more than 4 points (least squares mean changes from baseline were −6.82, −7.08, −6.54, and −5.71, for MF/F 400/10, MF/F 200/10, MF 400, and F 10, respectively). These changes were also significantly greater compared with placebo (−3.05, P ≤ 0.007, ). The proportion of subjects who achieved minimum clinically important difference changes from baseline of at least 4 points in SGRQ scores in the MF/F 400/10, MF/F 200/10, MF 400, F 10, and placebo groups was 52% (218/418), 53% (223/423), 53% (229/433), 51% (218/432), and 42% (173/414). The differences between proportions in all active treatment groups and the placebo group were statistically significant (P ≤ 0.011).
Figure 5 St George’s Respiratory Questionnaire total score change from baseline at week 26 endpoint.
Abbreviations: F, formoterol; MF, mometasone furoate; MF/F, mometasone furoate/formoterol fumarate fixed-dose combination formulation; SGRQ, St George’s Respiratory Questionnaire.
The proportion of COPD symptom-free nights was similar between groups at baseline (range 0.25–0.29); baseline assessment included symptoms over the week prior to the first dose of study medication. The proportion of COPD symptom-free nights over the 26-week treatment period increased in all treatment groups, with a least squares mean change from baseline of 0.15, 0.13, 0.14, 0.14, and 0.10 in the MF/F 400/10, MF/F 200/10, MF 400, F 10, and placebo groups, respectively. Compared with placebo, significantly greater improvements were seen in the MF/F 400/10, MF 400, and F 10 arms (P ≤ 0.033) but not in the MF/F 200/10 arm.
In the evaluation of pooled data for subjects with partly stable COPD over the last 8 weeks of treatment, no statistically significant differences were observed between treatments at the week 26 endpoint for the proportion of subjects with partly stable COPD. In the MF/F 400/10, MF/F 200/10, MF 400, F 10, and placebo groups, the proportions of subjects with partly stable COPD at endpoint were 39.5%, 44.3%, 38.1%, 42.0%, and 38.7%, respectively.
COPD exacerbations
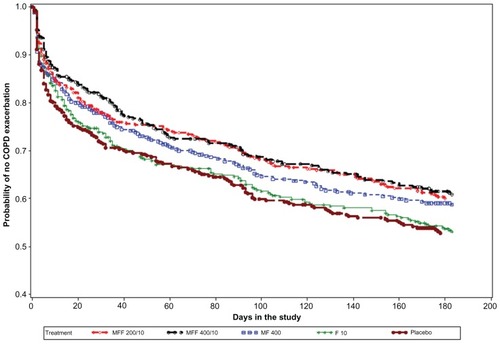
The percentage of subjects who experienced at least one mild, moderate, or severe COPD exacerbation across the 26-week treatment period in the MF/F 400/10, MF/F 200/10, MF 400, F 10, and placebo groups were 31.7%, 33.3%, 33.8%, 39.1%, and 39.4%, respectively. Based on the log-rank test for equality of survival curves (adjusted for smoking), MF/F 400/10, MF/F 200/10, and MF 400 were superior to placebo (P ≤ 0.038). MF/F 400/10 and MF/F 200/10 were also superior to F 10 (P ≤ 0.049, ). When only moderate and severe exacerbations were considered, the combination arms separated more distinctly from the other treatment arms. The percentage of subjects with moderate or severe COPD exacerbations was 12.1% with MF/F 400/10 and 20.7% with placebo. Based on adjusted survival curves, MF/F 400/10 was superior to MF 400, F 10, and placebo (P ≤ 0.030); MF/F 200/10 was superior only to F 10 and placebo (P ≤ 0.009).
Figure 6 Time-to-first mild, moderate or severe COPD exacerbation over the 26-week treatment period: Kaplan–Meier survival curves by treatment (all randomized subjects).
Pooled exposure-adjusted exacerbation rates showed that the incidence of exacerbations was lower in the MF/F and MF groups than it was in the formoterol group over the 52-week study period. The calculation of exposure-adjusted rates, expressed as events per patient-years, adjusts for the varying duration of exposure across treatments. The pooled exacerbation rates for the MF/F 400/10, MF/F 200/10, MF 400, and F 10 groups over the study period were 0.33, 0.34, 0.35, and 0.42 patient-years, respectively. Treatment advantages were marginally significant for MF/F 400/10 versus F 10 (P = 0.052) and MF/F 200/10 versus F 10 (P = 0.063) adjusting for smoking and study.
Using the Cox proportional hazard model, pooled hazard ratios for mild, moderate, or severe exacerbations with MF/F 400/10, MF/F 200/10, MF 400, and F 10 versus placebo were 0.761, 0.782, 0.825, and 0.972, respectively. The pooled hazard ratios for moderate or severe exacerbations with MF/F 400/10, MF/F 200/10, MF 400, and F 10 versus placebo were 0.571, 0.611, 0.828, and 0.957, respectively.
Safety
All four active treatments were well tolerated. During the 26-week treatment period, the percentage of subjects reporting any treatment-emergent adverse event was similar across the five treatment groups (31.8% with MF/F 200/10 to 36.2% with placebo). The most commonly reported treatment-emergent adverse events were headache, COPD, nasopharyngitis, upper respiratory tract infection, and hypertension (). COPD was reported as an adverse event if the subject had COPD symptoms or an exacerbation the investigator judged to have a clear temporal relationship with treatment administration. The incidence of COPD as a treatment-emergent adverse event was reported by 63 subjects (2.8%), ranging from 1.6% in the MF/F 200/10 group to 4.2% in the placebo group. During the treatment period, 164 subjects reported serious adverse events. COPD was by far the most frequent severe adverse event, reported by 15, 7, 12, 12, and 20 subjects in the MF/F 400/10, MF/F 200/10, MF 400, F 10, and placebo groups, respectively. Pooled results for treatment-emergent adverse events over the 52-week study period demonstrated that 17.0%–19.0% of subjects across the active treatment groups reported mild adverse events, whereas 15.4%–17.7% of subjects reported moderate adverse events. No more than 7.0% of subjects in any group reported severe treatment-emergent adverse events.
Table 5 Summary of treatment-emergent adverse events ≥2% incidence (all randomized subjects) during the treatment period
Treatment-related oral candidiasis, including esophageal and oropharyngeal, was reported by 19 subjects overall (0.8%) during the treatment period. Pneumonia, including lobar and viral pneumonia, was infrequent, occurring in nine (2.0%), five (1.1%), five (1.1%), six (1.3%), and three (0.7%) subjects in the MF/F 400/10, MF/F 200/10, MF 400, F 10, and placebo groups. During the 52-week study period, fewer than 6% of subjects in any treatment group reported any single treatment-emergent adverse event of any severity. Across the treatment groups, more subjects reported mild treatment-emergent adverse events than treatment-emergent adverse events of greater severity.
No abnormal, clinically relevant trends in laboratory values, vital signs, or electrocardiogram measurements were observed in the two studies. No clinically relevant trends or statistically significant treatment differences were observed for plasma cortisol levels, bone mineral density measurements, ocular changes, or forearm bruising.
Discussion
In this prospectively designed pooled analysis of two large, placebo-controlled Phase III studies in subjects with moderate to very severe COPD, significantly greater improvements in lung function were seen for twice-daily treatment using MF/F compared with placebo. Both dosing levels (MF/F 400/10 and MF/F 200/10) demonstrated significant improvements in both coprimary efficacy variables (morning predose/trough FEV1 and FEV1 AUC0–12 h) compared with placebo at all assessments from day 1 through week 26. In comparing the two combination regimens, the higher dose was associated with statistically greater improvements in both coprimary endpoints, demonstrating a significant dose-response relationship. Significant benefits in health status, night-time symptoms, and COPD exacerbation rates were also demonstrated with both MF/F doses. Both MF/F doses were well tolerated relative to their individual components and placebo.
Contribution of individual components
Although inhaled corticosteroids are frequently prescribed to reduce symptoms, improve health status, and decrease exacerbations,Citation25 inhaled corticosteroid monotherapy is not recommended in COPD treatment guidelinesCitation1 and is also not approved for treatment of COPD in the US.Citation25 However, MF has been shown to improve lung function and health status and reduce exacerbations significantly over 1 year.Citation13
Our results show that MF contributes to the efficacy of MF/F, as demonstrated by significantly greater improvement in morning predose/trough FEV1 with MF/F 400/10 compared with F 10. These differences were significant at day 1 and continued through 26 weeks. Improvements were not significantly different between inhaled corticosteroid monotherapy, MF 400, and placebo at the week 13 primary endpoint, but were significant at all other endpoints. Likewise, significantly greater improvements in FEV1 AUC0–12 h for MF/F 200/10 and 400/10 compared with F 10 across all time points from week 1 through week 26 demonstrate the contribution of MF to the combination.
LABAs are recommended for long-term management of COPD, and patients treated with formoterol, an approved COPD monotherapy in the US, demonstrate rapid onset of bronchodilation and sustained improvements in lung function with twice-daily dosing.Citation4,Citation26,Citation27 Our results show that formoterol contributes to the efficacy of combination therapy, as demonstrated by the significantly greater improvement in FEV1 AUC0–12 h with MF/F 400/10 compared with MF 400. Improvements in FEV1 AUC0–12 h were also significantly greater with F 10 than with placebo.
Health status, assessed using the SGRQ, was significantly improved with both combination doses. SGRQ has been widely utilized to assess health status in inhaled corticosteroid-LABA combination trials.Citation11,Citation18,Citation19,Citation28–Citation30 In studies up to 1 year, a change from baseline of more than 4 points has been reported with some combination regimensCitation18,Citation19 but not others.Citation18,Citation19,Citation29 Rennard et al and Tashkin et al compared budesonide/formoterol 160/9 μg twice daily with budesonide/formoterol 320/9 μg twice daily. In both studies, the change from baseline was >4 points in the lower-dose groups but <4 points in the higher-dose groups. In the current analysis, both MF/F doses resulted in clinically relevant improvements (>4 point change from baseline); however, the lower-dose group additionally demonstrated a change of >4 points compared with placebo. Effects such as these were not seen with higher-dose and lower-dose combinations in other studies,Citation11,Citation18,Citation19,Citation28–Citation30 which did not evaluate MF/F. MF/F 400/10 also demonstrated a significant improvement over placebo in the proportion of COPD symptom-free nights; however, the lower-dose combination did not show a significant difference over placebo.
The primary modes of action of LABAs and inhaled corticosteroids are distinct, namely bronchodilation and suppression of airway inflammation.Citation31 However, the following potential synergistic effects between the two classes have been suggested based on in vitro studies: inhaled corticosteroids enhance β2-adrenoceptor expression, which may prevent development of tolerance to β2-agonists with prolonged use; and LABAs amplify the anti-inflammatory effects of corticosteroids by accelerating nuclear translocation of the glucocorticoid receptor complex and enhancing transcription and expression of steroid-inducible genes in proinflammatory cells.Citation32,Citation33 Our analysis supports a synergistic effect of mometasone and formoterol on lung function. Based on morning predose/trough FEV1 measurements, MF/F 400/10 had a treatment effect (change from baseline) of 104 mL at the primary endpoint (13 weeks), which was greater than the summed treatment effects of MF and F (26 mL and 26 mL, respectively). Similarly, for standardized FEV1 AUC0–12 h, the treatment effect of MF/F 400/10 (172 mL) was greater than the sum of the treatment effects of MF 400 and F 10 (87 mL and 54 mL, respectively).
Although these trials were not designed to compare the efficacy of the two MF/F combination doses, the results clearly suggest greater benefits with MF/F 400/10 than with MF/F 200/10. Improvements in lung function (morning predose/trough FEV1 and FEV1 AUC0–12 h) were significantly greater with MF/F 400/10 compared with MF/F 200/10 at the week 13 endpoint, and were numerically greater at all assessment times.
Dose-dependent responses were also seen in the percentage of subjects who developed exacerbations of COPD symptoms and in reductions in night-time symptoms, although the differences between the higher-dose and lower-dose groups were not statistically significant.
Pooled exposure-adjusted exacerbation rates were lower in the MF/F groups than they were in the formoterol group, although the differences were marginally significant. In addition, compared with placebo, pooled hazard ratios for mild, moderate, or severe exacerbations were reduced 19% more in the MF/F 200/10 group and 21% more in the MF/F 400/10 group than they were in the formoterol group. Furthermore, pooled hazard ratios for moderate or severe exacerbations were reduced 35% more in the MF/F 200/10 group and 39% more in the MF/F 400/10 group than they were in the formoterol group.
Safety
MF/F was well tolerated at both dosing levels. The occurrence of total adverse events and treatment-related adverse events was similar across active treatment groups. A dose-related increase in the risk of pneumonia has been associated with the use of inhaled corticosteroid-containing regimens in COPD patients.Citation34,Citation35 We found that the incidence of pneumonia was low overall, although slightly higher in the MF/F 400/10 group than in other groups. Other studies have also reported an increase in oral candidiasis in inhaled corticosteroid-LABA trials.Citation16,Citation18 Oral candidiasis was very infrequent, with similar rates seen in the combination arms compared with non-MF-containing arms (F 10 and placebo). There was also an absence of any significant demonstrable adverse effect on bone density or ocular changes. While these data indicate MF/F was safe over the course of 1 year, longer trials should be conducted to confirm long-term safety.
Conclusion
In this pooled analysis of two large, 1-year, placebo-controlled clinical trials, patients treated with the MF/F pressurized metered-dose inhaler combination demonstrated significant improvements in lung function, health status, and exacerbation rates at the two doses investigated. Although significant improvements were seen at both combination doses, the higher dose was significantly more effective in improving lung function. A dose-response effect was observed in the lung function measurements.
Disclosures
DPT has served as a consultant for AstraZeneca, Boehringer Ingelheim, Dey Laboratories, and Merck & Co, received honoraria from AstraZeneca, Boehringer Ingelheim, and Dey Laboratories, and received grants from Almirall, AstraZeneca, Boehringer Ingelheim, Dey Laboratories, Merck & Co, Novartis, Pf izer, Sepracor, and Forest Laboratories. DED has served as a consultant for Forest, Ikaria, and Merck, has received via the University of Kentucky research/grant support from AstraZeneca, Boehringer Ingelheim, Pfizer, Merck, and Novartis, and received honoraria from AstraZeneca, Boehringer Ingelheim, Forest, Merck, and Pfizer. EK has received consulting fees from Dey Laboratories, GlaxoSmithKline, MAP Pharma (AstraZeneca), and Sepracor (Sunovion), and speaking fees from AstraZeneca, GlaxoSmithKline, Merck, and Teva. TS, DG, BK, and HS are employees of Merck Sharp & Dohme Corp. This study was sponsored by Merck Sharp & Dohme Corp. Medical writing and editorial assistance was provided by Karl Torbey and Ken Kauffman, AdelphiEden Health Communications, New York, NY. This assistance was funded by Merck Sharp & Dohme Corp, a subsidiary of Merck & Co, Inc, Whitehouse Station, NJ. Editorial assistance was also provided by Jorge Moreno-Cantu, Global Scientific and Medical Publications, Office of the Chief Medical Officer, Merck Sharp & Dohme Corp.
References
- QaseemAWiltTJWeinbergerSEDiagnosis and management of stable chronic obstructive pulmonary disease: a clinical practice guideline update from the American college of physicians, American college of chest physicians, American thoracic society, and European respiratory societyAnn Intern Med201115517919121810710
- RabeKFHurdSAnzuetoAGlobal strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summaryAm J Respir Crit Care Med200717653255517507545
- CelliBRMacNeeWStandards for the diagnosis and treatment of patients with COPD: a summary of the ATS/ERS position paperEur Respir J20042393294615219010
- AalbersRAyresJBackerVFormoterol in patients with chronic obstructive pulmonary disease: a randomized, controlled, 3-month trialEur Respir J20021993694312030736
- MahlerDADonohueJFBarbeeRAEfficacy of salmeterol xinafoate in the treatment of COPDChest199911595796510208192
- CasaburiRMahlerDAJonesPWA long-term evaluation of once-daily inhaled tiotropium in chronic obstructive pulmonary diseaseEur Respir J20021921722411866001
- CelliBRThomasNEAndersonJAEffect of pharmacotherapy on rate of decline of lung function in chronic obstructive pulmonary disease: results from the TORCH studyAm J Respir Crit Care Med200817833233818511702
- TashkinDPCelliBSennSA 4-year trial of tiotropium in chronic obstructive pulmonary diseaseN Engl J Med20083591543155418836213
- CoteCPearleJLSharafkhanehASpangenthalSFaster onset of action of formoterol versus salmeterol in patients with chronic obstructive pulmonary disease: a multicenter, randomized studyPulm Pharmacol Ther200922444919071226
- PauwelsRALofdahlCGLaitinenLALong-term treatment with inhaled budesonide in persons with mild chronic obstructive pulmonary disease who continue smoking. European Respiratory Society Study on Chronic Obstructive Pulmonary DiseaseN Engl J Med19993401948195310379018
- BurgePSCalverleyPMJonesPWRandomised, double blind, placebo controlled study of fluticasone propionate in patients with moderate to severe chronic obstructive pulmonary disease: the ISOLDE trialBMJ20003201297130310807619
- PaggiaroPLDahleRBakranIMulticentre randomised placebo-controlled trial of inhaled fluticasone propionate in patients with chronic obstructive pulmonary disease. International COPD Study GroupLancet19983517737809519948
- CalverleyPMRennardSNelsonHSOne-year treatment with mometasone furoate in chronic obstructive pulmonary diseaseRespir Res200897319014549
- National Institute for Clinical ExcellenceManagement of chronic obstructive pulmonary disease in adults in primary and secondary careClinical Guideline6122010 Available from: http://guidance.nice.org.uk/CG101Accessed January 19, 2012
- MahlerDAWirePHorstmanDEffectiveness of fluticasone propionate and salmeterol combination delivered via the Diskus device in the treatment of chronic obstructive pulmonary diseaseAm J Respir Crit Care Med20021661084109112379552
- HananiaNADarkenPHorstmanDThe efficacy and safety of fluticasone propionate (250 microg)/salmeterol (50 microg) combined in the Diskus inhaler for the treatment of COPDChest200312483484312970006
- CalverleyPPauwelsRVestboJCombined salmeterol and fluticasone in the treatment of chronic obstructive pulmonary disease: a randomised controlled trialLancet200336144945612583942
- RennardSITashkinDPMcElhattanJEfficacy and tolerability of budesonide/formoterol in one hydrofluoroalkane pressurized metered-dose inhaler in patients with chronic obstructive pulmonary disease: results from a 1-year randomized controlled clinical trialDrugs20096954956519368417
- TashkinDPRennardSIMartinPEfficacy and safety of budesonide and formoterol in one pressurized metered-dose inhaler in patients with moderate to very severe chronic obstructive pulmonary disease: results of a 6-month randomized clinical trialDrugs2008681975200018778120
- CalverleyPMKunaPMonsoEBeclomethasone/formoterol in the management of COPD: a randomised controlled trialRespir Med20101041858186820965712
- CazzolaMPasquaFFerriLRapid onset of bronchodilation with formoterol/beclomethasone Modulite and formoterol/budesonide Turbuhaler as compared to formoterol alone in patients with COPDPulm Pharmacol Ther20112411812220816833
- MillerMRHankinsonJBrusascoVStandardisation of spirometryEur Respir J20052631933816055882
- JonesPWQuirkFHBaveystockCMLittlejohnsPA self-complete measure of health status for chronic airflow limitation. The St George’s Respiratory QuestionnaireAm Rev Respir Dis1992145132113271595997
- JonesPWSt George’s Respiratory Questionnaire: MCIDCOPD20052757917136966
- GartlehnerGHansenRACarsonSSLohrKNEfficacy and safety of inhaled corticosteroids in patients with COPD: a systematic review and meta-analysis of health outcomesAnn Fam Med2006425326216735528
- CazzolaMCentanniSRegordaCOnset of action of single doses of formoterol administered via Turbuhaler in patients with stable COPDPulm Pharmacol Ther200114414511162418
- DahlRGreefhorstLANowakDInhaled formoterol dry powder versus ipratropium bromide in chronic obstructive pulmonary diseaseAm J Respir Crit Care Med200116477878411549532
- CalverleyPMAndersonJACelliBSalmeterol and fluticasone propionate and survival in chronic obstructive pulmonary diseaseN Engl J Med200735677578917314337
- SzafranskiWCukierARamirezAEfficacy and safety of budesonide/formoterol in the management of chronic obstructive pulmonary diseaseEur Respir J200321748112570112
- JonesPWAndersonJACalverleyPMHealth status in the TORCH study of COPD: treatment efficacy and other determinants of changeRespir Res2011127121627828
- SinDDManSFMarciniukDDCan inhaled fluticasone alone or in combination with salmeterol reduce systemic inflammation in chronic obstructive pulmonary disease? Study protocol for a randomized controlled trial [NCT0120978]BMC Pulm Med20066316460562
- MakJCNishikawaMShirasakiHMiyayasuKBarnesPJProtective effects of a glucocorticoid on downregulation of pulmonary beta 2-adrenergic receptors in vivoJ Clin Invest199596991067615841
- MakJCHisadaTSalmonMBarnesPJChungKFGlucocorticoids reverse IL-1beta-induced impairment of beta-adrenoceptor-mediated relaxation and up-regulation of G-protein-coupled receptor kinasesBr J Pharmacol200213598799611861327
- DrummondMBDasenbrookECPitzMWMurphyDJFanEInhaled corticosteroids in patients with stable chronic obstructive pulmonary disease: a systematic review and meta-analysisJAMA20083002407241619033591
- SinghSLokeYKRisk of pneumonia associated with long-term use of inhaled corticosteroids in chronic obstructive pulmonary disease: a critical review and updateCurr Opin Pulm Med20101611812219926996