Abstract
Background
Ipratropium bromide/albuterol Respimat inhaler (CVT-R) was developed as an environmentally friendly alternative to ipratropium bromide/albuterol metered-dose inhaler (CVT-MDI), which uses a chlorofluorocarbon propellant.
Objective
The objective of this study was to evaluate patient satisfaction, device usage, and long-term safety of CVT-R compared to CVT-MDI, and to the simultaneous administration of ipratropium bromide hydrofluoroalkane (HFA; I) and albuterol HFA (A) metered-dose inhalers as dual monotherapies (I + A).
Design
This is a 48-week, open-label, randomized, active-controlled, parallel-group study (n = 470) comparing CVT-R to CVT-MDI and to I + A.
Participants
Patients were at least 40 years of age, diagnosed with chronic obstructive pulmonary disease (COPD), and current or exsmokers.
Interventions
Patients were randomized to receive: (1) CVT-R, one inhalation four times daily (QID); or (2) CVT-MDI, two inhalations QID; or (3) I + A two inhalations of each inhaler QID.
Main measures
Patient Satisfaction and Preference Questionnaire (PASAPQ) performance score (primary endpoint) and adverse events.
Key results
PASAPQ performance score was significantly higher (CVT-R versus CVT-MDI, 9.6; and CVT-R versus I + A, 6.2; both P < 0.001) when using CVT-R compared to CVT-MDI or I + A at all visits starting from week 3, while CVT-MDI and I + A treatment groups were similar. Time to first COPD exacerbation was slightly longer in the CVT-R group compared to the other treatment groups, although it did not reach statistical significance (CVT-R versus CVT-MDI, P = 0.57; CVT-R versus I + A, P = 0.22). Rates of withdrawal and patient refusal to continue treatment were lower in CVT-R compared with CVT-MDI and I + A groups (CVT-R versus CVT-MDI, P = 0.09; CVT-R versus I + A, P = 0.005). The percentage of patients reporting adverse events and serious adverse events was similar across all three treatment groups.
Conclusion
CVT-R is an effective, environmentally friendly inhaler that provides patients with a high level of user satisfaction and may positively impact clinical outcomes while having no adverse impacts on patients using the device.
Background
Chronic obstructive pulmonary disease (COPD) guidelines recommend that bronchodilator therapies for COPD should be delivered via inhalation.Citation1 Four types of inhalers have been developed for the delivery of inhaled pharmacotherapy: nebulizers, pressurized metered-dose inhalers (pMDIs), dry powder inhalers, and low-velocity mist inhalers. Each inhaled delivery system impacts drug deposition in the lungs and has advantages and disadvantages. Correct inhaler usage is essential for optimal outcomes, with up to 70% of patients not using prescribed inhalers correctly,Citation2–Citation6 and up to 90% of patients with asthma or COPD making at least one critical error preventing effective medication delivery during MDI use.Citation7,Citation8 Even among health care professionals – whether physicians, residents, medical students, pharmacists, nurses, or respiratory therapists – studies have shown that practitioner skill in using inhalation devices ranges widely and is, overall, inadequate.Citation9
The Respimat (Boehringer Ingelheim Pharma GmbH and Co, KG, Ingelheim, Germany) inhaler is a novel, propellant-free low-velocity mist inhaler, relying on a mechanical spring-driven micro-pump to generate a slow-moving cloud of medication from an aqueous solution. Ipratropium bromide/albuterol delivered by a Respimat inhaler (CVT-R) is designed so that a single puff of CVT-R provides a similar dose-equivalent to two puffs of the currently available ipratropium bromide/albuterol metered-dose inhaler (CVT-MDI) using a chlorofluorocarbon (CFC) propellant for medication delivery. With the worldwide phase-out of CFCs, CVT-R was developed as an environmentally friendly alternative to CVT-MDI and has recently been approved by the US Food and Drug Administration (this device does not require and should not be used with a spacer). CVT-R has previously been shown in a 12-week study to be comparable to CVT-MDI with regard to bronchodilator efficacy and safety.Citation7,Citation10
Objective
The purpose of this study was to evaluate patient satisfaction, device usage, and the long-term safety of CVT-R. Using a three-arm study, CVT-R was compared to CVT-MDI and to ipratropium bromide (I) and albuterol (A) administered as dual monotherapies (I + A). This study design (ie, all treatment arms receiving the same drugs at fixed-dose equivalents via different formulations or delivery combinations) allowed attention to be focused on the delivery devices; the open-label design allowed for the comparison of the devices where blinding was not practically feasible.
Design
This study was a Phase III, 1-year, three-treatment, open-label, randomized, active-controlled, parallel-group study. A screening visit was followed by a 3- to 4-week baseline run-in period during which all patients received CVT-MDI. Patients were then randomized to receive one of three treatments in an open-label manner for 48 weeks. All patients were provided with albuterol (ProAir® hydrofluoroalkaline (HFA), Teva Respiratory, LLC, Horsham, PA, USA) for as-needed use during the baseline and treatment periods in addition to the investigational treatments.
Participants
Patients included in the study were at least 40 years of age, diagnosed with COPD, a forced expiratory volume in 1 second (FEV1) ≤80% of predicted, and an FEV1/forced vital capacity (FVC) ratio of ≤70%, and current or exsmokers with a smoking history of ≥10 pack-years. Patient eligibility was confirmed by a complete medical history, physical examination, 12-lead electrocardiography, spirometry (pulmonary function test), and clinical laboratory tests. Permissible and nonpermissible concomitant medications are listed in the Supplementary materials.
The clinical trial protocol and the informed consent were reviewed, and received approval from local or central Institutional Review Boards prior to the start of the study. The constitution of each Institutional Review Board met the requirements of the International Conference on Harmonisation (ICH). The trial was carried out in compliance with the protocol, the principles laid down in the Declaration of Helsinki, in accordance with the ICH Harmonised Tripartite Guideline for Good Clinical Practice (GCP), and in accordance with applicable regulatory requirements. Prior to patient participation in the trial, written informed consent was obtained from each patient (or the patient’s legally accepted representative) according to the ICH GCP and in accordance with the regulatory and legal requirements of the participating country. A signed copy of the informed consent and any additional patient information was given to each patient or the patient’s legally accepted representative.
Interventions
Patients were randomized to receive:
CVT-R, one inhalation four times daily (QID) (inhalation mist spray using the Respimat device with each actuation delivering 20 mcg ipratropium bromide [monohydrate] and 100 mcg albuterol from the mouthpiece); or
CVT-MDI, two inhalations QID (inhalation spray using a pMDI with a CFC propellant with each actuation delivering 18 mcg of ipratropium bromide and 103 mcg of albuterol sulfate [equivalent to 90 mcg albuterol base] from the mouthpiece); or
I + A, two inhalations of each inhaler QID (each inhalation spray using a pMDI with each actuation of I delivering 17 mcg of ipratropium bromide from the mouthpiece and each actuation of A delivering 108 mcg albuterol sulfate [90 mcg albuterol base] from the mouthpiece).
Information on randomization can be found in the Supplementary materials. Study medication was supplied by Boehringer Ingelheim Pharmaceuticals, Inc (Ingelheim, Germany).
Study treatments were not blinded to the patient or study center. However, all in-house handling of data was conducted in a blinded fashion.
Detailed written instructions and training for the use of the CVT-MDI and CVT-R inhalers were given to the patient at visit 1 and visit 2, respectively. Patients were instructed on how to prepare the inhaler for use (including inserting the cartridge into the inhaler and priming the unit) and using the CVT-R inhaler. For CVT-MDIs, patients were retrained as necessary on the correct priming technique in preparation for use, and use and care of each MDI. At all subsequent visits (visits 3–6), the investigator or qualified study personnel observed the inhalation procedure and reinforced the correct inhalation technique. Additionally, routine phone calls were made between visits to patients as a safety check, as well as to assess their understanding of proper inhalation and device utilization.
Main measures
The Patient Satisfaction and Preference Questionnaire (PASAPQ) is a self-administered instrument developed by experts in psychometric testing and validated to measure respiratory inhalation device satisfaction and preference in patients with asthma and COPD. The PASAPQ used in this trial contained 15 questions. The first 13 questions contained the performance domain (seven questions), the convenience domain (six questions), and the total score domain (all 13 questions). Question 14 asked for overall satisfaction with the device used in the study, and question 15 asked for willingness to continue with the device used in the study. The first 14 questions had Likert-type response options of 1 (very dissatisfied) to 7 (very satisfied); question 15 asked for responses between 0 and 100, with 0 indicating not willing to continue using the trial device and 100 indicating definitely willing to continue. The performance domain, convenience domains, and the total PASAPQ score have each been shown to independently correlate with patient satisfaction, with the highest correlation to patient satisfaction occurring with the PASAPQ performance domain.Citation11
As this study was not of a crossover design, patient comparison of two different study devices was not possible, and the PASAPQ administered for this study was modified to eliminate the standalone question asking for patient preference between the two devices. The lack of this question does not impact the satisfaction portion of the PASAPQ, including the performance and convenience domain scores, but could impact on the total PASAPQ score. The primary endpoint for this study was the PASAPQ performance domain score.
This PASAPQ was administered at visit 2 (prior to randomization), and at each treatment visit throughout the study. Patients randomized to I + A with the combination of two inhalers were asked to respond to the PASAPQ questions as if this was a single inhalation device/treatment. Secondary endpoints for the study included the PASAPQ overall satisfaction score, FEV1 and FVC changes from baseline pulmonary function tests, COPD exacerbations, and rescue medication use. A COPD exacerbation was defined as “a complex of lower respiratory events/symptoms (increase or new onset) related to the underlying COPD, with a duration of 3 days or more, requiring a change in treatment” where a “complex of lower respiratory events/symptoms” comprised at least two of the following: shortness of breath, sputum production (volume), occurrence of purulent sputum, cough, wheezing, or chest tightness. Other endpoints and medication restrictions are discussed in the Supplementary materials. Adverse events, regardless of causality, were recorded at each visit.
Treatment usage and compliance was assessed using a Daily Diary Card, in which study participants were required to enter the number of puffs of study medication taken, as well as the amount of rescue medication used. Compliance was calculated from the total number of puffs of study medication recorded as having been taken by the patient during the 2 weeks prior to each clinic visit divided by the number of days (with nonmissing data) for each patient.
Statistical analyses
Clinical data and statistical analyses were evaluated within the validated working environment, “Clinical Data Analysis and Reporting Environment,” and included processing and analyses with SAS® (version 9.2, SAS Institute Inc, Cary, NC, USA). The statistical design was a restricted maximum likelihood-based mixed-effect model repeated measure model and was used for comparisons of treatment groups for the performance domain score from the PASAPQ over time, including week 48. The fixed effects of treatment, test-day visit, treatment by test-day interaction, as well as the baseline PASAPQ scores, and baseline by treatment interaction were included in the mixed-effect model repeated measure model to adjust for the estimate of mean scores over time. All analyses were performed on the full analysis set consisting of all randomized patients who were documented to have taken at least one dose of trial medication. All analyses in this trial were descriptive and exploratory. All randomized and treated patients were included in the safety analysis. All safety data were displayed and analyzed using descriptive statistical methods. No inferential statistical analysis was planned for safety comparisons. Adverse events were coded using the MedDRA® (Medical Dictionary for Regulatory Activities) coding dictionary.
The sample size for this trial was based on regulatory requirements for safety and patient acceptability assessments of drugs intended for long-term treatment. To identify any adverse events with an incidence rate of at least 2%, a sample size of 150 patients per treatment group gave at least a 95% chance to observe at least one patient with that adverse event. This sample size (150 patients per treatment group) was able to detect an eight-point difference in the performance domain score between the CVT-R group and the CVT-MDI group, or between the CVT-R group and the I + A group with more than 90% power and using a two-sided 5% significance level, assuming a common standard deviation of 20 (two sample t-test with equal numbers from nQuery Advisor 6.01; Statistical Solutions, Saugus, MA, USA). A total of 600 patients (200 patients in each treatment group) were to be enrolled to ensure that 150 patients were randomized in each treatment group.
Key results
This study was conducted in 55 sites in the USA with a total of 688 patients enrolled, and 470 patients randomized (). The three treatment groups were comparable with respect to baseline demographics ().
Table A1 Permitted medications and medication restrictions
Table 1 Summary of study patient demographics
Figure 1 Study population.
Abbreviations: N, number; CVT-R, ipratropium bromide/albuterol Respimat inhaler; CVT-MDI, ipratropium bromide/albuterol metered-dose inhaler; I+A, ipratropium bromide and albuterol metered dose inhalers delivered as dual monotherapies; AE, adverse event.
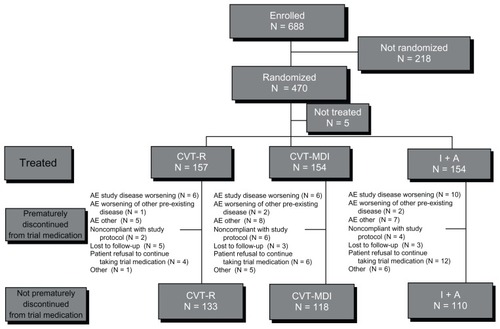
At the time of informed consent, 63% (n = 293) of the patients used pulmonary medications: inhaled short-acting beta agonists (45%), inhaled corticosteroids (33%), inhaled long-acting beta agonists (25%), inhaled short-acting anticholinergic agents (14%), inhaled long-acting anticholinergic agents (13%), and oxygen (6%). The CVT-R group had a slightly higher percentage of patients (68%, n = 107) taking pulmonary medications compared to the other two treatment groups (59% for CVT-MDI and 62% for I + A). Concomitant diagnoses (preexisting illnesses) at randomization were observed in 99% (n = 459) of study patients and were balanced across treatment groups. There was a slightly higher frequency of patients in the CVT-R treatment group (47%) having respiratory disorders other than COPD (eg, allergic rhinitis, and so on) compared to CVT-MDI (34%) and I + A (38%).
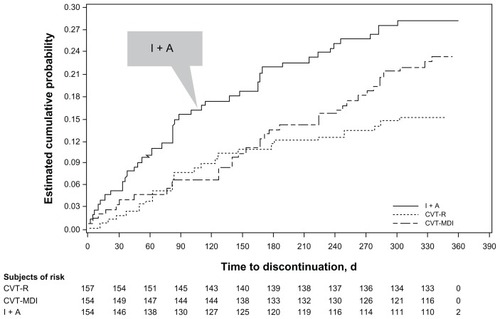
In total, 78% (n = 361) of patients completed the study (85% CVT-R; 77% CVT-MDI; 71% I + A). The CVT-R treatment group had lower and later withdrawal rates from the study compared to CVT-MDI and I + A. A total of 24 (15.3%), 36 (23.4%), and 44 (28.6%) patients prematurely discontinued treatment in CVT-R, CVT-MDI, and I + A groups, respectively (CVT-R versus CVT-MDI, hazard ratio [HR] 0.644, P = 0.095, CI: 0.384–1.079; CVT-R versus I + A, HR 0.487, P = 0.005, CI: 0.296–0.801). For I + A, the withdrawal curve diverges from the other two treatment groups from the beginning of the study period and remains divergent throughout the study (). Withdrawal curves for CVT-R and CVT-MDI were similar through 24 weeks and then diverged in the latter portion of the 48-week trial. Of the patients actively participating at the end of the trial (week 48), 95% achieved compliance in the “80% to 120%” range with CVT-R, 92% with CVT-MDI, and 92% with I + A.
Figure 2 Kaplan–Meier curves of time to discontinuation.
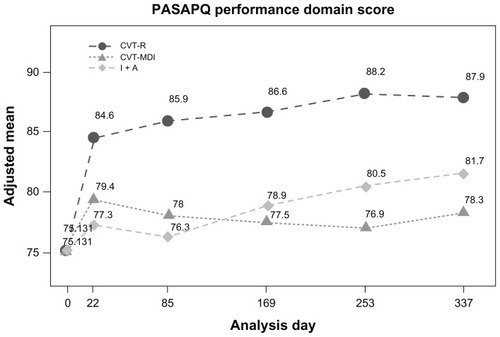
Differences in mean performance domain scores of the PASAPQ at week 48 (adjusted for baseline performance domain scores) were 9.6 for CVT-R versus CVT-MDI (P < 0.0001) and 6.2 for CVT-R versus I + A (P < 0.0001) (). No differences in PASAPQ performance scores were observed when comparing CVT-MDI to I + A at any of the on-treatment visits during the study. Of note, the CVT-R treatment group had an absolute increase in the mean PASAPQ performance scores (unadjusted for baseline) of 13.5 compared to baseline scores (when patients were using CVT-MDI).
Figure 3 Adjusted mean of PASAPQ performance domain score time profile.
Total PASAPQ scores were consistently higher for CVT-R compared to the other two treatment groups. For CVT-R compared to CVT-MDI, statistically significant differences were observed at week 24 (P < 0.001) and week 36 (P < 0.001), while statistically significant differences favoring CVT-R compared to I + A were present at each time point throughout the study (week 3, P = 0.025; week 12, P < 0.0001; week 24, P = 0.005; week 36, P < 0.001; week 48, P = 0.012).
Postbronchodilator increases in FEV1 and FVC following treatment were observed in all three treatment groups at each visit following study drug administration, with no clinically significant differences noted between any study arm at any time during the study (peak increases in FEV1 from baseline at week 48 of 0.22 L, 0.17 L, and 0.23 L for CVT-R, CVT-MDI, and I + A, respectively; CVT-R versus CVT-MDI, P = 0.03; CVT-R versus I + A, P = 0.88). Percent FEV1 increases from predicted values were 7.7%, 6.5%, and 8.0%, respectively. There were no differences in trough spirometry values among the three treatment groups throughout the study period.
A total of 124 (26.7%) patients had at least one exacerbation during the study (39, 41, and 44 patients in the CVT-R, CVT-MDI, and I + A groups, respectively). Most patients who had COPD exacerbation had only one (total = 96; 27, 32, and 37 for the CVT-R, CVT-MDI, and I + A groups, respectively). Twenty-one patients had two exacerbations, 21 patients had three exacerbations, five patients had four exacerbations, and two patients had four or more exacerbations throughout the 48-week study period. There were a total of 59, 50, and 53 COPD exacerbation events in the CVT-R, CVT-MDI, and I + A treatment groups, respectively. Overall, 24 (4.9%) patients had COPD exacerbations leading to hospitalization (ten, six, and eight for CVT-R, CVT-MDI, and I + A, respectively).
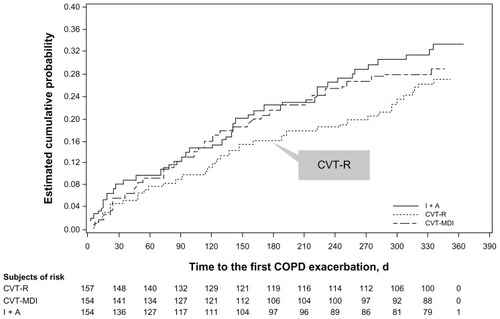
Time to first COPD exacerbation (TTFE) was not statistically different between the three treatment groups (CVT-R versus CVT-MDI, P = 0.57; CVT-R versus I + A, P = 0.22), although TTFE was numerically longer in the CVT-R group compared to the CVT-MDI and I + A treatment groups (). There were no differences among the three treatment groups for TTFE leading to hospitalization (CVT-R versus CVT-MDI, P = 0.35; CVT-R versus I + A, P = 0.79) or for exposure-adjusted event rates (event rate too low for statistical analysis).
Figure 4 Kaplan-Meier curves for time to first COPD exacerbation.
The number of puffs of rescue medication used was similar across the treatment groups (at week 48: CVT-R, 2.1; CVT-MDI, 1.8, P = 0.76; I + A, 1.7, P = 0.38), with treatment differences versus CVT-R ranging from −0.1 to 0.2 mean puffs (except for a single statistically significant higher use of rescue medication on day 169 for CVT-R versus I + A, P = 0.016).
Safety
All patients randomized to the trial and receiving at least one dose of study medication were included in the safety analysis. The increased retention of subjects in the CVT-R group resulted in a greater exposure to study treatment in this group compared to the other groups; however, this did not result in an increase in adverse events. The overall incidence of adverse events was comparable among the treatment groups.
A total of 72% (n = 335) of study patients reported an adverse event (). The percentage of patients with adverse events leading to discontinuation was higher in the I + A group (12.3%) compared with the CVT-R (7.0%) and CVT-MDI (9.7%) groups. The percentage of patients with serious adverse events was similar in all three treatment groups (14.6% CVT-R, 13.0% CVT-MDI, 16.2% I + A); however, the percentage of patients with fatal adverse events was higher in the I + A group (2.6%) compared to CVT-R (0.6%) and CVT-MDI (1.3%). Causes of death were as follows: CVT-R (n = 1), respiratory failure (no autopsy performed and no additional information is available); CVT-MDI (n = 2), severe sepsis and unknown cause; I + A (n = 4), metastatic pancreatic adenocarcinoma, cardiac arrhythmia, non-small-cell lung cancer stage 4 with metastasis to brain and liver, and myocardial infarction/coronary artery disease/perfusion defects/emphysema. All fatalities were considered by the investigators to be “not related” to the study drug.
Table 2 Frequency of patients (N, %) with adverse events occurring with incidence in preferred term greater than or equal to 3% by treatment, primary system organ class, and preferred term
Patients in the I + A group experienced more vomiting, while patients in the CVT-R group experienced a higher frequency of cough, chest pain, and musculoskeletal chest pain. All incidences of chest pain were considered to be noncardiac by the investigators. Adverse events consistent with anticholinergic (3.0%) or beta-agonist (14.0%) class effects were similar across all treatment groups.
Discussion
The objective of this study was to evaluate patient satisfaction, device usage, and long-term safety of CVT-R compared to CVT-MDI and the free combination of I + A. The results showed that there was greater patient satisfaction with CVT-R compared to CVT-MDI and to I + A, as evidenced by significantly higher PASAPQ performance scores (a difference of 9.6 at week 48 from baseline in the CVT-R versus the CVT-MDI group, and 6.2 in the CVT-R versus the I + A group). Kozma et alCitation11 have stated that for the performance domain, a difference of about ten points is needed to observe a medium effect, where the difference refers to a comparison between devices, either used concurrently or in sequence. Our results support a previous short-term study suggesting that patients prefer CVT-R to CVT-MDI.Citation10
Although high withdrawal rates can be detrimental to data analysis in long-term studies, insight into patient satisfaction, preferences, and acceptance of a medicine – and in this case, a device – can be gained from patient decisions to withdraw from a study. Patient preference for CVT-R is suggested by the lower withdrawal rates with CVT-R compared to the other treatment groups.
Interestingly, the switch from CVT-MDI to CVT-R or to I + A at study randomization did provide a form of crossover to an alternative device in this study. The comparison of absolute changes in PASAPQ performance scores during study run-in while using CVT-MDI to scores on treatment with CVT-R in the subjects randomized to CVT-R suggest a significant increase in patient satisfaction on the CVT-R compared to the CVT-MDI.
Similar outcomes for lung function, COPD exacerbations, and rescue medication usage in the three treatment groups were expected, as the same drugs with same dose equivalents were used in all three study arms. The divergence in TTFE was not expected. Although not statistically significant, the numerical difference in TTFE for CVT-R compared to the other treatment arms is interesting. This raises a question of whether device acceptance could impact on medication adherence and lead to improved clinical outcomes. Ultimately, the question of any improvement in TTFE based on delivery system requires further investigation.
This study also provides additional support for the long-term safety of CVT-R and confirms that CVT-R has a similar safety profile compared to CVT-MDI over 1 year.Citation7 Safety findings for CVT-R were comparable to CVT-MDI and to I + A, despite those patients receiving CVT-R having lower discontinuation rates and higher overall treatment exposures. Two adverse events were slightly higher in the CVT-R group: cough and chest pain. The higher reported cough rates in those receiving CVT-R may be due to the longer duration of spray and deeper particle penetration using the Respimat device. In fact, the overall differences between CVT-R and CVT-MDI may be due to the longer duration of the spray (1.5 seconds versus 0.15–0.36 seconds) and slower mean velocity (0.8 m/second versus 2.0–8.4 m/second) with CVT-R compared to CVT-MDI.Citation12 These two characteristics allow patients the time to coordinate actuation with inhalation more effectively. The reports of chest pain with CVT-R were nonspecific, noncardiac, and felt by the investigators to be “not related” to the use of study medication.
Study limitations
A modified version of the PASAPQ questionnaire was used in this trial, deleting a question asking patients to select a preferred device, because the trial did not employ a crossover design, and patients only used one device during the study. Importantly, the performance and convenience domains, as well as the total satisfaction question within the PASAPQ have been validated as standalone measurements correlating with patient satisfaction, and these domains and questions were not altered for this study.
Conclusion
CVT-R was superior to CVT-MDI and to the free combination I + A for patient satisfaction and delivery device usage. All three treatments were safe and well tolerated. In addition to showing improved patient satisfaction with and acceptance of CVT-R compared to CVT-MDI, this study confirms previous studies showing the safety and efficacy of CVT-R. Importantly, this device does not require and should not be used with a spacer, which is an added benefit as many patients do not carry a spacer with them when out of the home. Whether improved delivery of medication, improved patient satisfaction, and adherence to therapy based on a delivery device such as the Respimat can impact on other key COPD outcomes, such as TTFE, requires further investigation.
Supplementary materials
Rescue medication and additional treatments
Administration of albuterol as rescue medication was allowed at any time during the study. A different brand of albuterol HFA (other than the study medication) was provided for rescue, and the number of puffs used for rescue was to be recorded by the patient in the Daily Diary Card.
The following medications were allowed for control of acute exacerbations during the treatment period:
PRN albuterol inhalation aerosol (MDI) (provided by BIPI and its use to be recorded on the Patient Daily Diary Card).
Temporary increases in the dose of theophylline preparations of up to 7 days each were allowed during the study. If the increases or additions occurred prior to pulmonary function testing days, the testing was to be postponed for 2 days or to a maximum of 7 days after the last increased or additional dose was given.
Addition of oral steroids or temporary increases in the dose of steroids up to 7 days was allowed during the study. Pulmonary function testing was not to occur within 7 days of the last administered dose in the case of a steroid increase or addition. Pulmonary function testing was to be postponed up to 14 days to meet this restriction.
The use of antibiotics was not restricted and was to be used as medically necessary for exacerbations and other infections.
If an exacerbation or an upper respiratory tract infection/lower respiratory tract infection (URTI/LRTI) occurred anytime during the 3–4 week baseline period, the period was to be extended (up to 8 weeks) until the patient was stable enough to be randomized at Visit 2. If a second exacerbation or URTI/LRTI occurred during this period, the patient was excluded from the study.
The permitted medications and medication restrictions are outlined in the .
The following medications (other than the study medications) were not allowed during the baseline period or the treatment period.
Short-acting anticholinergic drugs including ATROVENT Inhalation Aerosol and ATROVENT Inhalation Solution by oral inhalation and for use in treating the common cold, ATROVENT Nasal Spray 0.06%.
Additional COMBIVENT Inhalation Aerosol or combination ipratropium bromide/albuterol solution for nebulization.
Oral beta-adrenergics or long acting beta-adrenergics such as salmeterol (Serevent™) and formoterol (Oxis®, Foradil®).
Short acting beta agonist other than the provided albuterol MDI.
Long-acting anticholinergic (Spiriva®). At least a 4-week washout during the baseline period was required for Spiriva.
Note: For patients using combination inhaled corticosteroids/long-acting beta adrenergics, therapies were to be switched to the inhaled corticosteroid monoproduct at Visit 1. The monoproduct inhaled corticosteroid did not need to be the same product, but was to be an equivalent dose. This monoproduct was to be used during the 3–4 week baseline period and continued throughout the study as appropriate. Albuterol MDI was to be used as additional PRN therapy.
Randomization
The order of assignment of the treatments was randomized. BI generated the randomization schedule and prepared the randomization. Prometrika LLC provided randomization services to the sites. Eligible patients were randomized to treatment at Visit 2. Their randomization number was also maintained within the eCRF. Each patient received open-label treatment for 48 weeks.
Secondary and other endpoints
Secondary endpoints:
Overall satisfaction score from the PASAPQ measured
Adverse events (AEs)
Rescue medication use (albuterol)
Physician’s Global Evaluation (PGE)
COPD Exacerbations reported as adverse events
FEV1 and forced vital capacity (FVC) change from baseline.
Other endpoints:
Dropout rates
Convenience domain score from the PASAPQ
Total score from the PASAPQ
Question (Q15) response to “willingness to continue” from the PASAPQ
Total score for the PASAPQ.
Acknowledgments
This study is supported by Boehringer Ingelheim. Under the authors’ conceptual direction, medical writing assistance for this paper was provided by Mary Gabb, MS of American Healthcare Communications; medical writing assistance was supported by Boehringer Ingelheim. Gary T Ferguson, MD, takes responsibility for the integrity of the work as a whole, from its inception to publication. The data from this trial were presented as a poster at the Society of General Internal Medicine meeting [Ferguson GT, Ghafouri M, Dai L, Dunn L. Acceptance of combivent Respimat inhalation spray in COPD patients. The 35th Annual Society of General Internal Medicine (SGIM) meeting; 2012 May 9–12; Orlando, Florida, 154 (Poster)]. ClinicalTrials.gov Identifier: NCT1019694. Combivent Respimat 1-year Safety Study in Patients with Chronic Obstructive Pulmonary Disease: http://clinicaltrials.gov/ct2/show/NCT1019694?term=NCT1019694&rank=1.
Disclosure
The authors report no conflicts of interest in this work. Gary Ferguson has received research funding, served as a consultant and a member of a speakers bureau for Boehringer Ingelheim. Luyan Dai is an employee of Boehringer Ingelheim. Mo Ghafouri is an employee of Boehringer Ingelheim. Leonard Dunn reports no conflicts of interest.
References
- Global Initiative for Chronic Obstructive Lung DiseasesGlobal strategy for the diagnosis, management, and prevention of COPD [webpage on the Internet]; 2011 [updated Dec 2011]. Available from: http://www.goldcopd.orgAccessed October 1, 2012
- FinkJBHodderRAdherence and inhaler devices in COPDRespir Ther2011612833
- VinckenWDekhuijzenPRBarnesPfor ADMIT GroupThe ADMIT series – Issues in inhalation therapy. How to choose inhaler devices for the treatment of COPDPrim Care Respir J2010191102019890594
- WieshammerSDreyhauptJDry powder inhalers in asthma and COPD: which factors determine the frequency of handling errors? A study of Aerosolizer, Discus, Handihaler and TurbuhalerChest2007132Suppl 4479s
- van BeerendonkIMestersIMuddeANTanTDAssessment of the inhalation technique in outpatients with asthma or chronic obstructive pulmonary disease using a metered-dose inhaler or dry powder deviceJ Asthma19983532732799661680
- BourbeauJBartlettSJPatient adherence in COPDThorax200863983183818728206
- EpsteinSWManningCPAshleyMJCoreyPNSurvey of the clinical use of pressurized aerosol inhalersCan Med Assoc J19791207813816427689
- HartertTVWindomHHPeeblesSFreidhoffLRTogiasAInadequate outpatient medical therapy for patients with asthma admitted to two urban hospitalsAm J Med199610043863948610724
- SelfTHArnoldLBCzosnowskiLMSwansonJMSwansonHInadequate skill of healthcare professionals in using asthma inhalation devicesJ Asthma200744859359817943567
- ZuwallackRDe SalvoMCKaelinTfor Combivent Respimat Inhaler Study GroupEfficacy and safety of ipratropium bromide/albuterol delivered via Respimat inhaler versus MDIRespir Med201010481179118820172704
- KozmaCMSlatonTLMonzBUHodderRReesePRDevelopment and validation of a patient satisfaction and preference questionnaire for inhalation devicesTreat Respir Med200541415215725049
- HochrainerDHölzHKreherCScaffidiLSpallekMWachtelHComparison of the aerosol velocity and spray duration of Respimat Soft Mist inhaler and pressurized metered dose inhalersJ Aerosol Med200518327328216181002